
薬学部の学生さんや製薬企業の若手の皆さんに、医薬品の開発がどのように進められたのかを知っていただくために、医薬品の研究開発経験者の視点で、審査報告書の中から要点を探ってみようと思い立ち、このブログを立ち上げました。ブログの書き方が不慣れですが、これからポリッシュアップしていきますので、どうぞよろしくお願いいたします! [第3回]
今回深掘りするトシリズマブ(ヒト化抗ヒトIL-6レセプターモノクローナル抗体)は、2005年に「キャッスルマン病」に関する効能・効果で承認されて以降、2008年に「関節リウマチ」及び「若年性特発性関節炎」、さらには2019年に「腫瘍特異的T細胞輸注療法に伴うサイトカイン放出症候群」及び「成人スチル病」に関する効能・効果が追加承認されており、本年1月に以下のような追加承認がなされました。
- 効能・効果:SARS-CoV-2)による肺炎(ただし、酸素投与を要する場合に限る)
- 用法・用量:通常、成人には、副腎皮質ステロイド薬との併用において、トシリズマブ(遺伝子組換え)として1回8mg/kgを点滴静注する。症状が改善しない場合には、初回投与終了から8時間以上の間隔をあけて、トシリズマブ(遺伝子組換え)として8mg/kgを更に1回追加投与できる。
製薬企業にとって、1つの医薬品の適応症が増えていくことはビジネス上大変有益です。年々売上を上げている本剤は適応症拡大のプロセスを知るうえで好事例と言えます。
適応症拡大が進む医薬品の審査プロセスを知るために、審査報告書を深掘りしてみましょう!
トシリズマブ(遺伝子組換え)アクテムラ点滴静注用80mg、200mg、400mgの審査報告書はこちら⇒450045000_21900AMX01337_A100_1.pdf (pmda.go.jp)
起原又は発見の経緯及び外国における使用状況に関する資料等
- 本剤について:
- 本剤の有効成分であるトシリズマブは、大阪大学と中外製薬株式会社との共同研究により創製された免疫グロブリンG1サブクラスのヒト化抗IL-6受容体モノクローナル抗体である。
- 本邦において本剤は、
- 2005年4月にキャッスルマン病に関する効能・効果で承認された。
- 2008年4月に関節リウマチ、若年性特発性関節炎が追加承認された。
- 2019年3月に腫瘍特異的T細胞輸注療法に伴うサイトカイン放出症候群、成人スチル病に関する効能・効果が追加承認された。:この項目に記載されていませんが、本薬の皮下注製剤(効能・効果は、関節リウマチ、高安動脈炎、巨細胞性動脈炎)も承認されています。
- 対象疾患に関して:
- 新型コロナウイルス感染症はSARS-CoV-2による感染症であり、本邦においては2020年1月15日にSARS-CoV-2に感染した1例目の患者が確認され、2020年2月1日、新型コロナウイルス感染症が感染症の予防及び感染症の患者に対する医療に関する法律(感染症法)に基づく指定感染症及び検疫法に基づく検疫感染症に指定された。
- 2021年12月13日現在、本邦における感染者(PCR陽性)は1,728,689例、死亡は18,373例と報告されている。
- SARS-CoV-2による感染症では、発熱、咳嗽、倦怠感、呼吸困難、味覚障害等の症状が認められ、約20%は肺炎症状が増悪し、一部の患者ではIL-6を含む複数のサイトカインの発現亢進を特徴とする高熱の炎症状態により呼吸不全を起こすこと、IL-6の上昇がSARS-CoV-2による感染症患者における死亡の予後因子の一つであったことが報告されている。
- MERS-CoV及びSARS-CoVに感染した患者の主要な死因はサイトカイン放出症候群(CRS)であるとの報告がある。
- 今回の製造販売承認事項一部変更承認申請の根拠:
- 本剤は、IL-6受容体を介したシグナル伝達を阻害する薬剤であり、腫瘍特異的T細胞輸注療法に伴うサイトカイン放出症候群に対する有効性及び安全性が確認されていたこと等から、SARS-CoV-2による肺炎に対する治療効果を期待して、国内外において複数の臨床試験が実施された。
- 得られた試験成績等に基づき、SARS-CoV-2による肺炎に対する本剤の臨床的有用性が確認された。
- 今般、SARS-CoV-2による肺炎に係る効能・効果を追加する製造販売承認事項一部変更承認申請(2021年12月13日)が行われた。:承認されたのが2022年1月21日なので、約1カ月のスビート審査でした。
- 海外の状況:
- 欧州ではF. Hoffmann-La Roche社により、海外医師主導臨床試験であるRECOVERY試験を主要な試験成績として、2021年7月27日に本剤のSARS-CoV-2による肺炎に係る効能・効果を追加する申請が行われ、2021年12月7日に承認された。
- 2021年12月13日時点において3の国又は地域で承認されている。
- 米国では、2021年12月13日時点において、SARS-CoV-2による肺炎に係る効能・効果で承認されていないが、2021年6月24日に、Emergency Use Authorizationを得ている。:Emergency Use Authorization(緊急使用許可)とは、米国FDAが緊急時に未承認薬などの使用を許可したり、既承認薬の適応を拡大したりする制度のことです。
品質、非臨床薬理試験、非臨床薬物動態試験、毒性試験
「品質に関する資料及び機構における審査の概略」
本申請は新効能及び新用量に係るものであり、「品質に関する資料」は提出されていない。
品質に関する資料は、平成17年に提出された初回の審査報告書に記載されています。⇒審査報告2マスクfinal.doc (pmda.go.jp) この審査報告書から品質関連の情報を抜粋しました。
「原薬」
(1)セルバンクの調製
ヒト骨髄腫細胞株U266から粗精製したヒト可溶性IL-6レセプターに対するマウスモノクローナル抗体を産生するハイブリドーマ細胞株から調製された重鎖及び軽鎖を、ヒトIgGの可変領域に組込み、ヒトDNAライブラリーからクローニング定常領域とそれざれ結合させて構築した。
この遺伝子発現構成体をCHO細胞株に導入して発現し、ウシ胎児血清を含まない培地でMCB(マスター・セル・バンク)を樹立した後、WCB(ワーキング・セル・バンク)が調整された。セルバンクの更新方法は、現在のMCB及びWCB調製時と同様な方法でMCBより調製し、特性解析及び純度試験を実施して現在のMCB及びWCBと同等な成績であることを確認する。:最近はMCBを更新しないケースが増えてきているようですが、本剤がロングセラーになった現在、MCB更新を設定していたのは良かったのだろうと思います。
(2)製造方法
製造工程は、(黒塗りが多く詳細は不明ですが)WCB解凍⇒拡大培養⇒生産バイオリアクター培養⇒クロマトグラフィー⇒ウイルス不活性化⇒2種のクロマトグラフィー⇒ウイルスろ過、濃縮・限外ろ過⇒添加剤添加・バルクろ過・充填・保管工程。:CHO細胞を用いた標準的な製造工程です。
(3)外来性感染性物質の安全性評価
初代MCB、初代WCB、MCB、WCB及びCALに対して無菌試験、マイコプラズマ試験及びウイルス試験が実施された。製造工程中で初代MCBの調製時にFBS米国)、BSA(米国)、? (米国、カナダ)、ヒト血漿由来トランスフェリン、ブタインスリン等が使用されていたことから、製造工程のウイルス除去・不活化能が検討された。(?は黒塗りで不明)
(4)製造方法の変更
黒塗りのため詳細は不明ですが、製法Aから製法Bに変更されたようです。
- 製法A(第Ⅰ相試験、臨床薬理試験、第Ⅱ相試験の一部に使用)
- 製法B(第Ⅱ相試験の一部に使用)
製法A及び製法Bによる原液から製造された治験薬を用いた臨床試験における生物薬剤学的な解析結果より、生物薬剤学的な差異が認められなかったため、製法A と製法B による原薬は同等/同質であると結論されている。
(5)特性
- 構造:①アミノ酸組成、②N末端アミノ酸配列、③C末端アミノ酸配列、④ジスルフィド結合、⑤ペプチドマップ、⑥糖鎖マッピング、⑦糖組成
- 物理化学的性質:①分子量(MALDI-TOF-MS)、②電気泳動的性質(等電点電気泳動(IEF)、SDS-PAGE)、③紫外吸収スペクトル、④円二色性スペクトル、⑤溶解度及びクロマトグラフィー的性質(ゲルろ過クロマトグラフィー(GPC)及びIEC)
- 免疫学的性質:免疫グロブリンサブクラス及びヒトIL-6Rに対する解離定数
- 生物学的性質:①IL-6R結合活性、②?細胞の増殖阻害活性、③カニクイザルにおけるIL-6の作用の阻害活性(?は黒塗りで不明です)
(6)原薬の管理
原薬の規格及び試験方法:申請時には性状、確認試験(SDS-PAGE)、pH、純度試験(GPC)、
製剤試験及び定量法(GPC、IL-6R 結合活性( ?法))が設定され、その後他の純度試験が追加設定された。:現在、バイオテクノロジー応用医薬品では通常、上記に加えて、含量、微生物限度、宿主細胞由来DNA、HCP(宿主細胞由来たん白質)、無菌、電荷バリアント、力価等が求められています(?は黒塗りで不明です)。
原薬の安定性:長期保存試験(-50℃及び-80℃、24カ月)、加速試験(5℃、6カ月)及び苛酷試験[温度(40℃・3カ月、50℃・3カ月、60℃・3週間)、光(120万lux・hr及び総近紫外放射エネルギー200W・hr/m2)]が実施された。:温度設定に±が無いですね。温度逸脱があったらどう対応したんでしょうね。苛酷試験の温度設定が3つあり、かなり綿密な安定性試験だと思います。
「製剤」
(1)製剤の製造について
製剤は、原薬に安定化剤としてポリソルベート80及び白糖を添加し、pH調整剤及び注射用水を加えて調製したもので、用時生理食塩液により希釈する溶液注射剤である。
(2)製造方法
製造方法は、①凍結保存されていた原薬を融解したものを、②あらかじめ混合・ろ過しておいた添加物と混合した後希釈して薬液を調製し、③無菌ろ過後20mLのガラスバイアルに充填、ゴム栓を打栓して製する。:バイオテクノロジー応用医薬品で標準的な製造工程を経ています。
(3)製剤の管理
製剤の規格及び試験方法:申請時には性状、確認試験(SDS-PAGE)、pH、純度試験(GPC)、製剤試験及び定量法(GPC、IL-6R 結合活性(?1 法))が設定されている。原薬と同様、純度試験として?2が追加設定された。:現在では通常、上記に加えて、浸透圧、エンドトキシン、採取用量、不溶性異物、不溶性微粒子、無菌、電化バリアント、力価等が求められています(?1と?2は黒塗りになっており不明です)。
製剤の安定性:長期保存試験5℃(24カ月)、加速試験25℃(6カ月)、苛酷試験[40℃(3カ月)、50℃(3カ月)、60℃(3週間)] 、光安定性試験、倒立(5℃、3カ月)、振動(JIS Z0232法により60分間振動)が実施された。:設定温度に±が無いですね。温度逸脱があったらどう対応したんでしょうね。過酷試験の温度設定が3つあったり、倒立や振動が実施され、かなり綿密な安定性試験だと思います。
「非臨床薬物動態試験に関する資料及び機構における審査の概略」
本申請は新効能及び新用量に係るものであるが、「非臨床薬物動態試験に関する資料」は過去の承認時に評価済みであるとされ、新たな試験成績は提出されていない。
非臨床薬物動態試験に関する資料は、平成17年に提出された初回の審査報告書に記載されています。⇒審査報告2マスクfinal.doc (pmda.go.jp) この審査報告書から関連の情報を抜粋しました。
(1)吸収:以下の4試験を実施した。
- 雄性ラットに本薬の125I-標識体0.5、5 及び50mg/kgを単回静脈内投与
- 雄性カニクイザルに本薬の125I-標識体5mg/kgを単回静脈内投与。本薬は血漿中ではほとんどが未変化体として存在している。
- 雌性カニクイザルに本薬5mg/kgを単回静脈内投与
- 雄性カニクイザルに本薬5mg/kg を1週間に1回、8週間反復静脈内投与
(2)分布:以下の3試験を実施した。
- 雄性ラットに本薬の125I-標識体5mg/kgを単回静脈内投与
- 雄性カニクイザルに本薬の125I-標識体5mg/kgを単回静脈内投与
- 妊娠 20日の雌性カニクイザルに妊娠50日まで本薬2、10及び50mg/kg を1日1回、31回反復静脈内投与
(3)代謝:以下の試験を実施した。
- 雄性ラットに本薬の125I-標識体5mg/kgを単回静脈内投与
- 雄性カニクイザルに本薬の125I-標識体5mg/kgを単回静脈内投与
- ラット及びカニクイザルともに本薬は血漿中では主として未変化体として存在していること、また、カニクイザルでは血漿中本薬の一部はsIL-6Rと複合体を形成していることが示唆されたと考察した。
(4)排泄:以下の2試験を実施した。
- 雄性ラットに本薬の125I-標識体0.5、5及び50mg/kgを単回静脈内投与試験。本薬のほとんどは代謝されてから尿中に排泄される。
- 雄性カニクイザルに本薬の125I-標識体5mg/kgを単回静脈内投与。投与放射能の74.3%及び2.46%がそれぞれ尿及び糞中に排泄された。
- 本薬は未変化体としては尿中にほとんど排泄されない。
(5)薬物相互作用:薬物相互作用に関する検討は実施されていない。
「毒性試験に関する資料及び機構における審査の概略」
本申請は新効能及び新用量に係るものであり、「毒性試験に関する資料」は過去の承認時に評価済みであるとされ、新たな試験成績は提出されていない。
毒性に関する資料は、平成17年に提出された初回の審査報告書に記載されています。⇒審査報告2マスクfinal.doc (pmda.go.jp) この審査報告書から品質関連の情報を抜粋しました。
毒性に関する資料:以下の試験が実施された。
- 単回毒性試験:ラット、カニクイザル(いずれも静脈内投与)
- 反復投与毒性試験:ラットで2週(用量設定試験)及び4週、カニクイザルで2週(用量設定試験)を含む4週及び26週(いずれも静脈内投与)
- 生殖発生毒性試験:ラット受胎能及び着床までの初期胚発生に関する試験、ラット胚・胎児
- 発生への影響に関する試験、ウサギ胚・胎児発生への影響に関する試験、カニクイザル胚・胎児
- 発生への影響に関する試験(いずれも静脈内投与)
- 遺伝毒性試験:細菌を用いる復帰突然変異試験、ヒト末梢血リンパ球を用いる染色体異常試験
- がん原性試験:実施せず
生物薬剤学試験及び関連する分析法、臨床薬理試験に関する資料並びに機構における審査の概略
「生物薬剤学試験及び関連する分析法」
血清中本薬濃度は、酵素結合免疫吸着測定法(定量下限:0.1μg/mL)により測定された。
「臨床薬理試験」
評価資料として下記を提出した:
- SARS-CoV-2による肺炎患者を対象とした国内臨床試験(J-COVACTA試験)の成績
参考資料として下記を提出した:
- 外国人SARS-CoV-2による肺炎患者を対象とした海外第Ⅱ相試験(MARIPOSA試験)の成績
- 海外第Ⅲ相試験(COVACTA試験)の成績
国内第Ⅲ相試験(J-COVACTA試験〔2020年5月~2020年12月〕)
- 対象:18歳以上のSARS-CoV-2による肺炎患者(48例投与)
- 1回投与症例:本剤8mg/kg(最大800mg)を静脈内投与、41例
- 2回投与症例:初回投与後、臨床兆候若しくは症状が悪化若しくは改善しない場合に初回投与から8~24時間後に本剤8mg/kg(最大800mg)を1回追加投与、7例
- 血清中本薬濃度の薬物動態パラメータ(Cmax, AUCmf, t1/2, CL, MRT, Vd)を示した。
- 本剤投与後の全集団における投与後の血清中CRP及びIL-6濃度の推移を示した。
- CRP濃度の平均値は本剤投与後に低下した。
- IL-6濃度の平均値は本剤投与後に増加した後に低下が認められたが、一部の症例では7~14日目に上昇が認められた。
- 申請者によると、これらの症例に関しては、併用薬、臨床症状等の推移から、併用していた副腎皮質ステロイド薬が中止されたことが影響した可能性があるとのこと。
海外第Ⅲ相試験(COVACTA 試験〔2020 年4 月~2020 年7 月〕)
- 対象:18歳以上のSARS-CoV-2による肺炎患者
- プラセボ対照無作為化二重盲検並行群間比較試験
- 1回投与症例:本剤8mg/kg(最大800mg)を静脈内投与
- 2回投与症例:初回投与後、臨床兆候又は症状が悪化又は改善しない場合に初回投与から8~24 時間後に本剤8mg/kg(最大800mg)を1回追加投与
- 本剤群及びプラセボ群における血清中CRP 濃度推移を図に示した。:図によると、投与3、 7、14、21日目で本剤群の血清中CRP濃度はプラセボ群よりも低値を示した(有意差検定なし)。
PK の国内外差について
申請者は、国内臨床試験(J-COVACTA試験)及び海外臨床試験(MARIPOSA試験及びCOVACTA
試験)において、本剤を申請用法・用量で投与した際の血清中本薬濃度の推移を表に示し、得られた結果からSARS-CoV-2による肺炎患者における本剤のPK に明らかな民族差はないと考える旨、説明している。:表には、上記3試験の、国内外の血清中本薬濃度の推移(投与15分後から34日後まで)のみを列挙して、明確な民族差はないとしています。通常用いられるCmax、AUC、t1/2など主要なPKパラメータが示されていませんが、すでに国内外で使用されている本剤のPKに民族差がないことは分かっているので血清中濃度のみに留めたのだと思います。
「機構における審査の概略」
PMDAは、SARS-CoV-2による肺炎患者に対する本剤の有効性及び安全性の根拠に海外臨床試験成績を用いることについて、薬物動態の観点から特段の問題は示唆されていないと考える、と回答した。
臨床的有効性及び臨床的安全性に関する資料並びに機構における審査の概略
評価資料:国内臨床試験(第Ⅲ相試験、J-COVACTA試験)
参考資料:
- 海外医師主導臨床試験(RECOVERY試験)
- 海外第Ⅲ相試験(COVACTA試験)
- 海外第Ⅲ相試験(EMPACTA試験)
- 海外第Ⅲ相試験(REMDACTA試験)
「国内臨床試験」J-COVACTA試験〔2020年5月~2020年12月〕
- 対象:18歳以上のSARS-CoV-2による肺炎患者
- 目的:本剤の有効性、安全性等を検討する
- デザイン:非盲検非対照試験:唯一の評価資料の試験がsingle-armの非盲検非対象試験というのは異例ですが、コロナウイルス感染のパンデミックの真っただ中で開発された背景を考慮すると理解できます。
- 目標登録例数:10例(11施設)
- 選択基準
- いずれかの検体(呼吸器、血液、尿、便、その他の体液等)でPCR陽性が確認され、胸部X線又はCTスキャンにより確認されたSARS-CoV-2による肺炎で入院した患者
- SpO2が93%以下又はPaO2/FiO2が300mmHg未満
- 除外基準
- AST又はALTが基準値上限の10倍超:本薬など抗体医薬品は肝臓などの細網内皮系の細胞で代謝されるから
- 好中球数1,000/μL未満:本薬に免疫抑制作用があるから
- 血小板数50,000/μL未満:血小板はIL-6によって一部産生されるから
- 妊婦又は授乳婦
- 3カ月以内に経口免疫抑制薬又は免疫調整薬(本剤を含む)を投与された患者
- 用法・用量
- 標準治療併用下において本剤8mg/kg(最大800mg)を1時間かけて静脈内投与する、臨床症状が悪化又は改善しない場合は、初回投与から8~24時間後に1回追加投与可能。
- 結果
- 登録症例数:49例、うち48例全例が有効性解析対象集団及び安全性解析対象集団
- 一回投与患者:41/48例
- 二回投与患者:7/48例
- 中止例:7/48例、中止理由は死亡6例、被験者の申し出1例
- 有効性:主要評価項目である7段階順序尺度を用いて評価した投与28日後における臨床状態の結果
- 2段階以上の改善:36/48例(75.0%、95%CI[60.40, 86.36])
- 1段階以上の改善:39/48例(81.3%、95%CI[67.37, 91.05])
- 1段階以上の悪化:6/48例(12.5%、95%CI[4.73, 25.25])
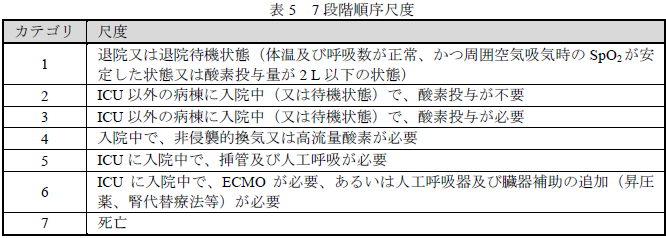
:7段階順序尺度は、本剤以外のCOVID-19の臨床試験でも使用されています。
- 安全性
- 有害事象:83.3%に認められ、10.0%以上に認められた事象は便秘18.8%、ALT増加、フィブリンDダイマー増加及び高尿酸血症各16.7%、AST増加14.6%、肝障害10.4%
- 重篤な有害事象:12.5%(〔COVID-19肺炎3例、COVID-19肺炎、細菌性肺炎、呼吸不全及び肺梗塞各1例(重複あり)〕)に認められたが、いずれも本剤との因果関係は否定された。
- 試験中止に至った有害事象(死亡例を除く):なし
- 副作用:31.3%
「海外医師主導臨床試験」RECOVERY試験〔2020年4月~(2021年3月29日データカットオフ)〕)(Lancet 2021; 397: 1637-45)
- 対象:18歳以上のSARS-CoV-2による肺炎患者
- 目的:本剤の有効性等を検討する
- デザイン:標準治療を対象とした非盲検無作為化並行群間比較試験
- 目標登録例数:4,000例(英国の131施設)、(本剤群:標準治療群=1:1)
- 用法・用量
- 本剤群:本剤の既承認用量(1回8mg/kg)を基本に、標準治療併用下で体重区分ごとに設定された用量(体重40kg以下:8mg/kg、体重40kg超65kg以下:400mg、体重65kg超90 kg以下:600mg、体重90kg超:800mg)を1時間以上かけて静脈内投与し、患者の状態の改善が認められない場合には、12~24時間後に1回追加投与可能とする。
- 標準治療群:実施中の標準治療(1回目の無作為化において割付けられた治療を含む)を継続する。
- 結果
- 登録症例数:本剤検討コホートの組入れ基準を満たした4,116例が無作為化された。
- 解析:無作為化された全例(本剤群2,022例、標準治療群2,094例)をITT集団とし、ITT集団が有効性検討対象とした。
- 有効性:主要評価項目とされた無作為化28日目までの全死亡の結果を表及び図に示した。
- 全死亡数:本剤群621/2022例(31%)、標準治療群729/2094例(35%)、ハザード比0.85、95%信頼区間[0.76, 0.94] 、p値0.0028
- 安全性:本剤に関連すると考えられる重篤な副作用として、外耳炎、黄色ブドウ菌血症及び肺膿瘍各1件が報告された。
「海外第Ⅲ相試験」COVACTA試験〔2020年4月~2020年7月〕
- 対象:18歳以上のSARS-CoV-2による肺炎患者
- 目的:本剤の有効性、安全性等を検討する
- デザイン:プラセボ対照無作為化二重盲検並行群間比較試験
- 目標登録例数:450例(本剤群:プラセボ群=2:1)
- 施設:62施設、9カ国(米国、カナダ、デンマーク、フランス、ドイツ、イタリア、オランダ、スペイン及び英国)
- 選択基準:国内臨床試験と同じ
- いずれかの検体(呼吸器、血液、尿、便、その他の体液等)でPCR陽性が確認され、胸部X線又はCTスキャンにより確認されたSARS-CoV-2による肺炎で入院した患者
- 室内気でSpO2が93%以下又はPaO2/FiO2が300 mmHg未満
- 除外基準:下記ラインマーカーの2項目以外国内臨床試験と同じ
- AST又はALTが基準値上限の10倍超
- 好中球数1,000/μL未満
- 血小板数50,000/μL未満
- 妊婦又は授乳婦
- 3カ月以内に経口免疫抑制薬又は免疫調整薬(本剤を含む)を投与された患者
- 活動性の細菌、真菌又はSARS-CoV-2以外のウイルス感染が疑われる患者
- 24時間以内の死亡が不可避と考えられる患者
- 用法・用量:国内臨床試験と同じ(除くプラセボ)
- 標準治療併用下において、本剤8mg/kg(最大800mg)又はプラセボを1時間かけて静脈内投与し、臨床症状が悪化又は改善しない場合は、初回投与から8~24時間後に1回追加投与可能とした。
- 結果
- 登録症例数:452例(本剤群301例、プラセボ群151例)が、地域及び侵襲的人工呼吸器管理実施の有無を層別因子として無作為化された。
- 解析:無作為化された452例のうち、438例に試験薬が投与され(本剤群294例、プラセボ群144例)、この438例がmITT集団とされ、mITT集団が有効性解析対象集団とされた。
- mITT集団のうち、プラセボ群に割り付けられた1例に本剤が投与され、安全性解析では本剤群として取り扱われたので、安全性解析対象集団は本剤群295例、プラセボ群143例であった。
- 一回投与患者:本剤230/295例(78.0%)、プラセボ100/143例(69.9%)
- 二回投与患者:本剤65/295例(22.0%)、プラセボ43/143例(30.1%)
- 中止例:本剤群35.4%(104/294例)、プラセボ群33.3%(48/144例)、中止理由は死亡(本剤群24.1%、プラセボ群25.0%)、追跡不能(本剤群7.8%、プラセボ群3.5%)、被験者の申し出(本剤群3.4%、プラセボ群2.7%)等
- 有効性:主要評価項目である試験薬投与開始28日目における7段階順序尺度を用いて評価した臨床状態の結果を表に示し、本剤群とプラセボ群との間に統計学的な有意差なし(p=0.36、有意水準両側5%、地域及び侵襲的人工呼吸器管理実施の有無を層別因子としたVan Elteren検定)。:国内臨床試験と同じ7段階順序尺度を用いてます。
- 安全性
- 有害事象:本剤群81.4%、プラセボ群82.5%に認められ、いずれかの群で3.0%以上に認められた事象は、COVID-19肺炎、尿路感染、急性腎障害、高血圧、便秘、下痢、肺炎、貧血、戯言、COVID-19、心房細動、不眠症
- 死亡に至った有害事象:
- 本剤群24.4%(〔COVID-19肺炎36例、COVID-19 13例、多臓器機能不全症候群5例、呼吸不全3例、敗血症性ショック、急性呼吸窮迫症候群、心停止及び後腹膜出血各2例、急性呼吸不全、肺塞栓症、出血性ショック、出血、凝血異常、卒中の出血性変化及び急性腎障害各1例〕)。多臓器機能不全症候群、後腹膜出血及び敗血症性ショック各1例)については試験薬との因果関係は否定されず。
- プラセボ群25.2%(〔COVID-19肺炎20例、呼吸不全3例、COVID-19、急性呼吸窮迫症候群及び急性呼吸不全各2例、敗血症性ショック、細菌性肺炎、肺塞栓症、誤嚥、肺硬化、心停止及び出血性ショック各1例〕)
- 重篤な有害事象:本剤群39.3%、プラセボ群44.8%に認められ、いずれかの群で1.0%以上に認められた事象を表にまとめた。
- 本剤群18例(好中球減少症4例、敗血症性ショック及び細菌性肺炎各3例、菌血症及び肺炎各2例、大腸菌性肺炎、シトロバクター検査陽性、細菌性敗血症、サイトメガロウイルス肝炎、エンテロバクター性肺炎、下部消化管出血、尿路感染、多臓器機能不全症候群、敗血症、後腹膜出血及び膵炎各1例(重複あり))
- プラセボ群13例(敗血症性ショック及び肺炎各3例、カンジダ感染、血小板減少症、ステノトロフォモナス感染、汎血球減少症、細菌性肺炎、菌血症、敗血症、胆嚢炎、呼吸不全、尿路性敗血症及びシュードモナス性敗血症各1例(重複あり))については試験薬との因果関係は否定されなかった。:プラセボとの因果関係が否定されないこともあります。
- 試験中止に至った有害事象(死亡例を除く):いずれの群においても認められず。
- 副作用:本剤群18.3%、プラセボ群18.2%
「海外第Ⅲ相試験」EMPACTA試験〔2020年5月~2020年9月〕
- 対象:18歳以上のSARS-CoV-2による肺炎患者
- 目的:本剤の有効性、安全性等を検討する
- デザイン:プラセボ対照無作為化二重盲検並行群間比較試験
- 目標登録例数:379例(本剤群:プラセボ群=2:1)
- 施設:42施設、6カ国(米国、メキシコ、ケニア、南アフリカ、ペルー及びブラジル)
- 選択基準:
- いずれかの検体(呼吸器、血液、尿、便、その他の体液等)でPCR陽性が確認され、胸部X線又はCTスキャンにより確認されたSARS-CoV-2による肺炎で入院した患者
- 室内気でSpO2が94%未満:この基準が国内臨床試験と異なる
- 除外基準:下記ラインマーカーの5項目が国内臨床試験と異なる
- 持続陽圧呼吸、二相性陽圧換気又は侵襲的人工呼吸器管理を要する患者
- AST又はALTが基準値上限の5倍超
- 好中球数1,000/μL未満
- 血小板数50,000/μL未満
- 妊婦又は授乳婦
- 3カ月以内に経口免疫抑制薬又は免疫調整薬(本剤を含む)を投与された患者
- 腸管穿孔歴のある患者
- 活動性の細菌,真菌又はSARS-CoV-2以外のウイルス感染が疑われる患者
- 24時間以内の死亡が不可避と考えられる患者
- 用法・用量:国内臨床試験と同じ(除くプラセボ)
- 標準治療併用下において、本剤8mg/kg(最大800mg)又はプラセボを1時間かけて静脈内投与し、臨床兆候又は症状が悪化又は改善しない場合は、初回投与から8~24時間後に1回追加投与可能とした。
- 結果
- 登録症例数:388例(本剤群259例、プラセボ群129例)が、国及び年齢(60歳以下又は60歳超)を層別因子として無作為化された。
- 解析:無作為化された388例のうち、377例に試験薬が投与され(本剤群249例、プラセボ群128例)、この377例がmITT集団とされ、mITT集団が有効性解析対象集団とされた。
mITT集団のうち、プラセボ群に割り付けられた1例に本剤が投与され、安全性解析では本剤群として取り扱われたので、安全性解析対象集団は本剤群250例、プラセボ群127例であった。 - 一回投与患者:本剤182/250例(72.80%)、プラセボ92/127例(72.4%)
- 二回投与患者:本剤68/250例(27.2%)、プラセボ35/127例(27.6%)
- 中止例:本剤群9.6%(24/249例)、プラセボ群10.2%(13/128例)、中止理由は死亡(本剤群9.6%、プラセボ群8.6%)等
- 有効性:主要評価項目である試験薬投与開始28日までの人工呼吸器使用又は死亡の結果を表に示し、本剤群とプラセボ群との間に統計学的な有意差が認められた(人工呼吸器使用又は死亡した患者数:本剤群29/249例(11.6%)、プラセボ群24/128例(18.8%)、ハザード比0.56、95%信頼区間0.33, 0.97、p=0.0360、有意水準両側5%、年齢を層別因子とした層別log-rank検定)。:国内臨床試験とは異なる評価項目を用いてます。
- 安全性
- 有害事象:本剤群50.8%、プラセボ群52.8%に認められ、いずれかの群で3.0%以上に認められた事象は、便秘(本剤群6.4%、プラセボ群3.1%、以下同順)、不安(6.0%、3.1%)、頭痛(3.2%、2.4%)、疲労(1.2%、3.9%)、急性腎障害(1.6%、3.1%)、高血糖(1.2%、3.1%)、肺炎(0.8%、3.1%)
- 死亡に至った有害事象:
- 本剤群11.2%(〔急性呼吸窮迫症候群5例、急性呼吸不全及び呼吸不全各4例、COVID-19肺炎、敗血症性ショック、心停止及び心肺停止各2例、COVID-19、ブドウ球菌性肺炎、急性心筋梗塞、脳幹卒中、脳血管発作、腸管穿孔及び多臓器機能不全症候群各1例〕)
- プラセボ群10.2%(〔COVID-19肺炎3例、急性呼吸不全及び呼吸不全各2例、急性呼吸窮迫症候群、呼吸窮迫、敗血症性ショック、細菌性肺炎、心房細動及び心筋梗各1例〕)に認められ、本剤群1例(ブドウ球菌性肺炎)については試験薬との因果関係は否定されず。:プラセボとの因果関係が否定されないこともあります。
- 重篤な有害事象:本剤群15.2%、プラセボ群19.7%に認められ、いずれかの群で1.0%以上に認められた事象は敗血症性ショック(本剤群2.0%、プラセボ群2.4%、以下同順)、急性呼吸窮迫症候群(2.0%、0.8%)、急性呼吸不全(1.6%、2.4%、呼吸不全(1.6%、1.6%)、肺塞栓症(1.2%、0.8%)、COVID-19肺炎(0.8%、2.4%)、急性腎障害(0.4%、2.4%)、肺炎(0、2.4%)及び細菌性肺炎(0、1.6%)であった。本剤群3例(菌血症、感染性胆嚢炎、医療機器関連感染及びブドウ球菌性肺炎各1例(重複あり))については試験薬との因果関係は否定されず。
- 試験中止に至った有害事象(死亡例を除く):いずれの群においても認められず。
- 副作用:本剤群12.8%、プラセボ群3.9%
「海外第Ⅲ相試験」REMDACTA試験〔2020年6月~2021年3月〕)
- 対象:12歳以上のSARS-CoV-2による肺炎患者
- 目的:本剤の有効性、安全性等を検討する
- デザイン:プラセボ対照無作為化二重盲検並行群間比較試験
- 目標登録例数:650例(本剤群:プラセボ群=2:1)
- 施設:53施設、4カ国(米国、ブラジル、ロシア及びスペイン)
- 選択基準:
- いずれかの検体(呼吸器、血液、尿、便、その他の体液等)でPCR陽性が確認され、胸部X線又はCTスキャンにより確認されたSARS-CoV-2による肺炎で入院した患者
- 室内気でSpO2超を維持するために6L/min超の酸素投与を要する患者:この基準が国内臨床試験と異なる
- 除外基準:下記ラインマーカーの6項目が国内臨床試験と異なる
- AST又はALTが基準値上限の5倍超
- 好中球数1,000/μL未満
- 血小板数50,000/μL未満
- 妊婦又は授乳婦
- 3カ月以内に経口免疫抑制薬又は免疫調整薬(本剤を含む)を投与された患者
- 2回超のレムデシビルを投与された患者
- 推定糸球体濾過量が30mL/mim未満又は血液透析中の患者
- 体重40kg未満
- 活動性の細菌,真菌又はSARS-CoV-2以外のウイルス感染が疑われる患者
- 24時間以内の死亡が不可避と考えられる患者
- 用法・用量:国内臨床試験ではレムデシビルは併用せず
- レムデシビル併用下において、本剤8mg/kg(最大800mg)又はプラセボを1時間かけて静脈内投与し、臨床兆候又は症状が悪化又は改善しない場合は、初回投与から8~24時間後に1回追加投与可能とした。
- レムデシビルは、1日目に200mgを、2~10日目に100mgを1日1回静脈内投与し、退院した場合は投与を中止する。
- 結果
- 登録症例数:649例(本剤群434例、プラセボ群215例)が、地域及び疾患の重症度を層別因子として無作為化された。
- 解析:無作為化された649例のうち、640例に試験薬が投与され(本剤群430例、プラセボ群210例)、この640例がmITT集団とされ、mITT集団を有効性解析対象集団とした。
- レムデシビル又は試験薬(本剤又はプラセボ)が投与された642例を安全性解析対象集団とした。
- 本剤群に割り付けられた症例のうち誤ってプラセボが投与された1例及び試験薬未投与でレムデシビルが投与された2例が安全性解析ではプラセボ群として取り扱われたので、安全性解析対象集団は本剤群429例、プラセボ群213例であった。
- 一回投与患者:本剤344/429例(80.2%)、プラセボ163/211例(77.3%)
- 二回投与患者:本剤85/429例(19.8%)、プラセボ48/211例(22.7%)
- 中止例:本剤群30.5%(131/430例)、プラセボ群33.3%(70/210例)、中止理由は死亡(本剤群22.3%、プラセボ群25.7%、以下同順)、追跡不能(5.3%、5.7%)、被験者からの申し出(2.1%、1.9%)等
- 有効性:主要評価項目である無作為化から28日目までの退院又は退院待機状態までの期間の結果を表と図に示し、両群間で統計学的な有意差は認められなかった(退院又は退院待機状態となった患者数:本剤群284/430例(66.0%)、プラセボ群141/210例(67.1%)、退院又は退院待機状態までの期間の中央値 [95%CI] (日):本剤群14.0 [12.0, 15.0] 、プラセボ群14.0 [11.0, 16.0] 、ハザード比0.56、95%信頼区間0.33, 0.97、p=0.7414、有意水準両側5%、地域及び疾患の重症度を層別因子とした層別log-rank検定)。:国内臨床試験とは異なる評価項目を用いてます。
- 安全性
- 有害事象:本剤群77.4%、プラセボ群71.8%に認められ、いずれかの群で3.0%以上に認められた事象は、便秘、急性腎障害、COVID-19肺炎、肺炎、トランスアミナーゼ上昇、尿路感染、敗血症ショック、低血圧、低カリウム血症、高血糖、不眠症、心房細動、悪心
- 死亡に至った有害事象:
- 本剤群22.6%(〔COVID-19肺炎35例、COVID-19 14例、敗血症性ショック13例、肺炎6例、敗血症5例、アシネトバクター性肺炎、ショック、出血性ショック、心原性ショック及び死亡各2例、出血性卒中、脳血管発作、塞栓性脳卒中、クレブシエラ菌性肺炎、肺敗血症、徐脈、上室性頻脈、腸管虚血、後腹膜出血、多臓器機能不全症候群、肺塞栓症、誤嚥性肺炎、気胸及び脳ヘルニア各1例〕)に認められ、うち敗血症性ショック3例、敗血症、肺炎、アシネトバクター性肺炎、クレブシエラ菌性肺炎、腸管虚血及び多臓器機能不全症候群各1例について試験薬との因果関係は否定されず。
- プラセボ群25.8%(〔COVID-19肺炎26例、COVID-19 8例、敗血症性ショック3例、敗血症、出血性卒中及び多臓器機能不全症候群各2例、肺炎、細菌性肺炎、尿路性敗血症、死亡、脳死、頭蓋内出血、低酸素性虚血性脳症、血行動態不安定、腸管虚血、大腸炎、肺塞栓症及び急性腎障害各1例〕)に認められ、うち敗血症性ショック及び尿路性敗血症各1例について試験薬との因果関係は否定されず。:プラセボとの因果関係が否定されないこともあります。
- 重篤な有害事象:本剤群32.9%、プラセボ群35.7%に認められ、いずれかの群で1.0%以上に認められた事象は、COVID-19肺炎(本剤群8.4%、プラセボ群12.7%、以下同順)、敗血症性ショック(5.4%、4.7%)、急性腎障害(4.9%、6.6%)、肺炎(4.9%、2.8%)、COVID-19(3.3%、3.8%)、敗血症(2.6%、2.8%)、気胸(1.6%、0.5%)、細菌性肺炎(1.2、1.9%)及び肺塞栓症(1.2、1.4%)であった。
- 本剤群34例(肺炎12例、敗血症性ショック7例、敗血症5例、尿路感染4例、細菌性肺炎、急性腎障害、細菌性肺炎及び肝損傷各2例、腎機能障害、腸管虚血、クレブシエラ菌性肺炎、気管気管支炎、多臓器機能不全症候群、エンテロバクター感染、カテーテル留置部位出血、ショック、細菌性敗血症、気胸、アシネトバクター性肺炎及び血小板数減少各1例(重複あり))については試験薬との因果関係は否定されず。
- プラセボ群16例(敗血症性ショック及び肺炎各4例、敗血症、気管気管支炎及び全身性カンジダ2例、尿路感染、エンテロバクター性菌血症、細菌性肺炎、ブドウ球菌感染、アシネトバクター性肺炎、肝酵素上昇、肝不全、尿路性敗血症、肺敗血症、急性肝炎、憩室穿孔、菌血症、医療機器関連敗血症及びクレブシエラ菌性肺炎各1例(重複あり)については試験薬との因果関係は否定されず。
- 試験中止に至った有害事象(死亡例を除く):本剤群0.5%(2/429例〔脳血管発作及び急性腎障害各1例〕)に認められ、いずれも試験薬との因果関係は否定された。
- 副作用:本剤群14.9%、プラセボ群12.7%
機構における審査の概略
「有効性及び臨床的位置づけについて」
申請者の説明は、
本剤の有効性について
- 主に酸素投与を要するSARS-CoV-2による肺炎患者を対象とし、全死亡の評価に対して一定の検出力を有すると考えられる症例数で実施されたRECOVERY試験において、
- 本剤投与により全死亡割合が低下する結果が得られている。
- 当該試験におけるベースライン時点の副腎皮質ステロイド薬併用有無別の結果において、標準治療群と比較した本剤群の無作為化後28日目までの全死亡のハザード比[95%CI]は、副腎皮質ステロイド薬併用あり及びなしの集団でそれぞれ0.79[0.70, 0.89]及び1.16[0.91, 1.48]であり、副腎皮質ステロイド薬併用なしの集団では1を上回った。
- RECOVERY試験、並びに申請者が実施したCOVACTA試験、EMPACTA試験及びREMDACTA試験における28日目までの全死亡の結果を表にまとめた。
- 海外3試験では、28日目までの全死亡割合について一貫した結果は得られていないが、いずれも検討した症例数が少なく全死亡に基づき本剤の有効性を評価する上で十分な検出力を有していないことに加えて、試験実施時期の違いにより標準治療の変化等があり、副腎皮質ステロイド薬を併用していた患者の割合が異なったことや組み入れられた患者の疾患重症度の違い等が影響した可能性があると考えている。
- WHOにおいて実施されたSARS-CoV-2による感染症の入院患者における全死亡とIL-6阻害薬投与との関連性を推定するためのメタアナリシス(27の無作為化比較試験)において(論文の引用)、
- 標準治療又はプラセボ投与した患者と比較したIL-6阻害薬を投与した患者の28日目までの全死亡のオッズ比[95%CI]は全体集団で0.86[0.79, 0.95]、ベースライン時副腎皮質ステロイド薬併用ありの集団で0.78[0.69, 0.88]、ベースライン時副腎皮質ステロイド薬併用なしの集団で1.09[0.91, 1.30]であった。
- このうち、本剤が用いられた19試験における当該オッズ比[95%CI]は、全体集団で0.83[0.74, 0.92]、ベースライン時副腎皮質ステロイド薬併用ありの集団で0.77[0.68, 0.87]、副腎皮質ステロイド薬併用なしの集団で1.06[0.85, 1.33]であり、副腎皮質ステロイド薬との併用で本剤投与により全死亡割合が低下することが示唆された。
- RECOVERY試験(本剤検討コホート)ではCRP値が75mg/L以上の患者が組み入れ対象であったが、このメタアナリシスでは、本剤を用いた14試験において28日目までの全死亡のオッズ比に対するCRP値の影響は示唆されていない。
本剤の臨床的位置づけについて
- 本邦のCOVID-19診療の手引きでは、本邦で承認されている薬剤として、レムデシビル、デキサメタゾン(副腎皮質ステロイド剤)などの記載があるが、酸素投与を要するSARS-CoV-2による肺炎患者に対する治療選択肢は限られている。
- これまでに、日本人SARS-CoV-2による肺炎患者を対象に本剤の有効性及び安全性を標準治療又はプラセボと比較した臨床試験は実施していないが、以下の点から、日本人患者に対する臨床的有用性について、外国人患者を対象とした臨床試験成績に基づき評価することは可能。
- SARS-CoV-2による感染症患者において認められる諸症状、重症化のリスク因子等に国・地域による明らかな違いはない。
- 海外3試験に参加した国・地域及びRECOVERY試験で本剤の評価が行われた英国ではSARS-CoV-2による感染症患者に対して重症度に応じ酸素投与、人工呼吸、ECMOによる管理、薬物治療として重症度に応じてカシリビマブ/イムデビマブ等の中和抗体薬、レムデシビル、副腎皮質ステロイド剤等の投与が行われ、加えて血栓症対策や合併症管理が実施されており、本邦とSARS-CoV-2による感染症の治療方針に著しい違いはない。
- 本剤の薬物動態を検討した国内外の臨床試験の結果から、SARS-CoV-2による肺炎患者における薬物動態に明らかな民族差はない。
- SARS-CoV-2による肺炎患者における本剤の安全性について、日本人患者において外国人患者と比較して新たな安全性上の懸念はない。
- 本剤は腫瘍特異的T細胞輸注療法に伴うサイトカイン放出症候群に対して国内外で承認されており、当該効能・効果の対象患者において日本人集団と外国人集団における有効性及び安全性に明らかな違いはない。
- RECOVERY試験の成績等に基づき、本剤の一定の有効性が確認されたこと、SARS-CoV-2による肺炎患者に対して本剤は忍容可能であったことから、副腎皮質ステロイド薬併用下において、本剤は、酸素投与を要するSARS-CoV-2による肺炎患者に対する治療選択肢の一つになり得る。
この説明に対してPMDAは、
- RECOVERY試験は通常の臨床試験と比較として収集する有効性、安全性等に係る情報が限定されており、試験で得られた各被験者に関する情報の量及び精度については限界があったと考えるが、SARS-CoV-2による感染症パンデミック下において大規模の無作為化比較試験を実施する上での方策として、一定の理解は可能である。
- RECOVERY試験は非盲検下で実施されているが、客観的な事象である全死亡に基づき有効性が評価されていることに加え、有効性解析対象集団が約4,000例と規模の大きい臨床試験であることも踏まえ、当該試験成績から全死亡の結果について一定の評価を行うことは可能と判断した。
- 以上の判断及び現時点で酸素投与を要するSARS-CoV-2による肺炎患者に対する治療法が限られている現状を踏まえ、本剤の有効性及び臨床的位置づけに関する申請者の説明は理解可能であり、対象患者は異なるものの、腫瘍特異的T細胞輸注療法に伴うサイトカイン放出症候群に対する本剤の有効性及び安全性の国内外差に関する知見や、RECOVERY試験成績に基づくEUでの承認取得等を考慮すると、副腎皮質ステロイド薬併用下において、本剤は、酸素投与を要するSARS-CoV-2による肺炎患者に対する治療選択肢の一つになり得る。
以上の機構の判断については、専門協議において議論したい、回答した。
安全性について
申請者の説明は、
(1)安全性プロファイル
SARS-CoV-2による肺炎患者対象の海外3試験併合データ及び国内臨床試験に基づき、本剤の安全性について検討した。これらの臨床試験における安全性の概要を表にまとめた。:海外3試験併合データとは、海外第Ⅲ相試験(COVACTA試験、EMPACTA試験、REMDACTA試験)のデータを併合したもの。
- 海外3試験併合データの全体集団のプラセボ群(483例)と比較して本剤群(974例)において、
- 発現割合が2.0%以上高かった有害事象は高血圧(本剤群3.6%、プラセボ群1.4%、以下同順)
- 発現割合が0.5%以上高かった死亡に至った有害事象はCOVID-19(2.9%、2.1%)、敗血症性ショック(1.7%、1.0%)
- 発現割合が0.5%以上高かった重篤な有害事象はCOVID-19(3.0%、2.1%)、細菌性敗血症及び後腹膜出血(0.5%、0)
- 発現割合が2.0%以上高かったGrade3以上の有害事象はなかった。
- 海外3試験併合データの全体集団(974例)における本剤群と国内臨床試験(48例)の比較において、
- 国内臨床試験で発現割合が5.0%以上高かった有害事象は便秘(国内18.8%、海外9.0%、以下同順)、ALT増加(16.7%、2.3%)、フィブリンDダイマー増加(16.7%、1.0%)、高尿酸血症(16.7%、0)、AST増加(14.6%、1.6%)、肝障害(10.4%、0)、肝機能異常(6.3%、0.6%)、皮膚剝脱(8.3%、0)、皮膚炎(6.3%、0.2%)
- 国内臨床試験で2例以上に認められ、海外3試験併合データの全体集団における本剤群より発現割合が2.0%以上高かったGrade3以上の有害事象、死亡に至った有害事象及び重篤な有害事象なし。
- 海外3試験併合データの全体集団における本剤群と比較して国内臨床試験においてALT増加、フィブリンDダイマー増加等の臨床検査値に関連する事象の発現頻度が高い傾向が認められた要因の一つとして、海外3試験と比較して国内臨床試験で検査頻度が高かったことが影響した可能性が考えられる。
既承認の効能・効果の承認審査において本剤投与時の重要な特定されたリスクとされた事象(重篤な感染症、腸管穿孔、アナフィラキシー等の重篤な過敏症、B型肝炎ウイルスの再活性化、好中球減少・白血球減少・無顆粒球症、血小板減少、肝機能障害、間質性肺炎)の発現状況を表にまとめた。:ここに挙げられたすべての事象について、ベースライン時副腎皮質ステロイド薬併用の有無にかかわらず、本剤投与群はプラセボ投与群よりも高い頻度を示した。ただし、国内臨床試験では、腸管穿孔、アナフィラキシー等の重篤な過敏症及び間質性肺炎はなかった。
この説明に対してPMDAは、
海外3試験併合データにおいてプラセボ群と比較して本剤群で重篤な事象の発現割合が高く、既承認の効能・効果の承認審査において本剤投与時の重要な特定されたリスクとされていない出血について、重点的に検討を行った。
(2)出血
申請者の説明は、
臨床試験における出血関連事象の発現状況を表にまとめた。
- 海外3試験併合データにおいて認められた死亡に至った出血は、
- 本剤群(974例)で後腹膜出血及び出血性ショック各3例、出血性卒中、卒中の出血性変化、出血及び凝血異常各1例
- プラセボ群(483例)で出血性卒中2例、頭蓋内出血及び出血性ショック各1例
- 本剤群の後腹膜出血1例は試験薬との因果関係は否定されず。
- 海外3試験併合データにおいて認められた重篤な出血は、
- 本剤群(974例)で後腹膜出血5例、出血性ショック4例、メレナ、血腫、卒中の出血性変化及びカテーテル留置部位出血各2例、下部消化管出血、口腔内出血、直腸出血、動脈出血、出血、出血性卒中、脳出血、失血性貧血、凝血異常、播種性血管内凝固、鼻出血、血胸及び処置後出血各1例
- プラセボ群(483例)で胃腸出血、出血性卒中、頭蓋内出血及び咽頭出血各2例、メレナ、腹壁血腫、腸間膜血腫、上部消化管出血、出血性ショック、血腫、くも膜下出血、失血性貧血及びカテーテル留置部位出血各1例
- 本剤群の下部消化管出血、後腹膜出血及びカテーテル留置部位出血各1例は試験薬との因果関係は否定されず。
- SARS-CoV-2による肺炎患者では血栓症の合併頻度が高く、各国・地域のガイドラインにおいても抗血栓薬の投与が標準的治療の1つに含まれており、海外3試験併合データでは、本剤群の94.7%、プラセボ群の95.0%、国内臨床試験では70.8%で抗血栓薬が使用されていた。臨床試験において重篤な出血が認められた患者では、全例で該当する事象の発現時又は発現前に抗血栓薬が使用されており、抗血栓薬を使用していなかった集団において認められた出血は海外3試験併合データの本剤群血便1例、国内臨床試験のフィブリンDダイマー増加及び喀血各1例であった。
- 安全性データベースにおいて、SARS-CoV-2による肺炎患者に本剤を投与した際の出血に関連する事象は国内症例14例(重篤な事象13件、非重篤な事象13件)、海外症例89例(重篤な事象96件、非重篤な事象8件)で報告されているが、本剤との因果関連が明確な事象は認められず。
以上を踏まえると、現時点において本剤投与による出血について注意喚起する必要性は乏しい。
この説明に対してPMDAは、
- 申請者の説明を了承し、現時点において本剤投与による明らかな出血関連リスクは示唆されていないと判断した。:本剤との因果関係が否定できなかった死亡例1例と重篤な出血3例は、どのように解釈されたのでしょうか?
- 以上の検討を踏まえ、
- 得られた臨床試験成績から本剤投与時の安全性プロファイルが既承認の効能・効果に対する使用時と明らかに異なる傾向は認められていない。
- 国内臨床試験における検討例数が限られていること等から、国内外差の検討に限界はあるが、得られている臨床試験成績から日本人患者において外国人患者と比較して新たな安全性上の懸念は認められていない。
以上の機構の判断については、専門協議において議論したい、と回答した。
効能・効果について
申請者の説明は、
RECOVERY試験等に基づき、酸素投与を要するSARS-CoV-2による肺炎患者に対して本剤の一定の有効性が示され、当該患者に対して本剤は忍容可能と考えたことから、
- 効能・効果は「SARS-CoV-2による肺炎(ただし、酸素投与を要する患者に限る)」と設定した。
- 本剤の投与対象を明確化するため、効能・効果に関連する注意において「酸素投与、人工呼吸管理又は体外式膜型人工肺(ECMO)導入を要する患者を対象に入院下で投与を行うこと。」と注意喚起する。
この説明に対してPMDAは、
提出された資料、「有効性及び臨床的位置づけについて」及び「安全性について」の検討を踏まえ、申請者が示す効能・効果を設定すること及び効能・効果に関連する注意を行うことは適切と考える。
以上の機構の判断については、専門協議において議論したい、と回答した。
用法・用量について
申請者の説明は、
- 海外3試験及び国内臨床試験における本剤の用法・用量は、既承認の腫瘍特異的T細胞輸注療法に伴うサイトカイン放出症候群に対する体重30kg以上の患者の用法・用量及び中国における使用経験に基づき、
- 本剤8mg/kg(最大800mg)を1時間かけて静脈内投与する。
- 臨床症状が悪化又は改善しない場合は、初回投与終了8~24時間後に1回追加投与が可能とする。
- 臨床試験の成績から、酸素投与を要するSARS-CoV-2による肺炎患者に対して、副腎皮質ステロイド薬との併用により本剤の一定の有効性は示され、忍容可能と考えられたので、
- 用法・用量は「通常、成人には、副腎皮質ステロイド薬との併用において、トシリズマブとして1回8mg/kgを点滴静注する。症状が改善しない場合には、初回投与終了から8時間以上の間隔をあけて、トシリズマブとして8mg/kgを更に1回追加投与できる。」と設定した。
- 酸素投与を要するSARS-CoV-2による感染症患者に対する治療薬として承認されているレムデシビル及びバリシチニブとの併用については、
- レムデシビルについて、海外3試験の併合データにおいて本剤群の974例中587例、プラセボ群の483例中297例、国内臨床試験の48例中23例で併用されており、その結果、レムデシビルを併用することによる安全性上の特段の懸念を認めず。
- バリシチニブについて、JAK阻害薬と本剤の作用機序は一部重複することから、国内臨床試験及び海外3試験では、バリシチニブを含むJAK阻害薬の併用を禁止した。SARS-CoV-2による肺炎患者を対象とした臨床試験においてバリシチニブの併用投与の情報が極めて限られている(1例のみ)ことから、バリシチニブとの併用は推奨されない。
この説明に対してPMDAは、
- 申請者の説明を了承し、記載整備した上で、以下のように設定することが適切である。
- <用法・用量>通常、成人には、副腎皮質ステロイド薬との併用において、トシリズマブ(遺伝子組換え)として1回8mg/kgを点滴静注する。症状が改善しない場合には、初回投与終了から8時間以上の間隔をあけて、トシリズマブ(遺伝子組換え)として8 mg/kgを1回追加投与できる。
- バリシチニブとの併用について、臨床試験ではJAK阻害薬は併用禁止とされており、SARS-CoV-2による肺炎患者における併用使用の経験は極めて限られていることを踏まえると、添付文書において併用時の有効性及び安全性は確立していない旨を注意喚起する必要がある。
以上の機構の判断については、専門協議において議論したい。
まとめ
2019年末に中国・武漢で感染が拡大し始めた新型コロナウイルスは、2020年1月には日本で1例目の感染が、2021年12月には18,373例の死亡が報告されました。
本剤のSARS-CoV-2に対する第Ⅲ相試験は、2020年5月に開始され(終了は同年12月)、この試験成績を評価資料、海外医師主導治験1本及び海外第Ⅲ相試験3本の成績を参考資料として、2021年12月13日に「SARS-CoV-2による肺炎」の適応症追加が薬事申請されました。
承認されたのは2022年1月21日なので、年末年始を挟んでいることを勘案すると、約1カ月という超短期間で承認審査が行われています。
新型コロナウイルス感染症が指定感染症及び検疫感染症に指定され、有効な治療薬が大至急で求められるコロナ禍、開発担当者は(おそらく)テレワークで、自身も感染の不安を持ちながら、申請書類の作成とPMDAとのコミュニケーションを短期間で遂行されたのだろうと感心しました。
さて、このブログの冒頭で、「製薬企業にとって、1つの医薬品の適応症が増えていくことはビジネス上大変有益です」と述べました。
薬価は、現在では毎年見直され(かつては2年に1度)、原則的には減額されています。
新薬の研究開発には膨大な費用がかかります。
- 2016年の論文によると、1つの新薬の承認及び承認後のR&D費用を合算すると、28.7億ドルに達すると報告されており(Journal of Health Economics 47:20, 2016)、年々増加しています。
したがって、新薬開発をおこなう各製薬企業にとって、手持ちの商品の売上げ最大化が重要課題の一つとなっています。
アクテムラの薬価も例外なく減額されています。
- 例えば、点滴静注用400mgの2012年4月以前の薬価は117,459円でしたが、2022年4月には56,073円と半値以下に減額されています。
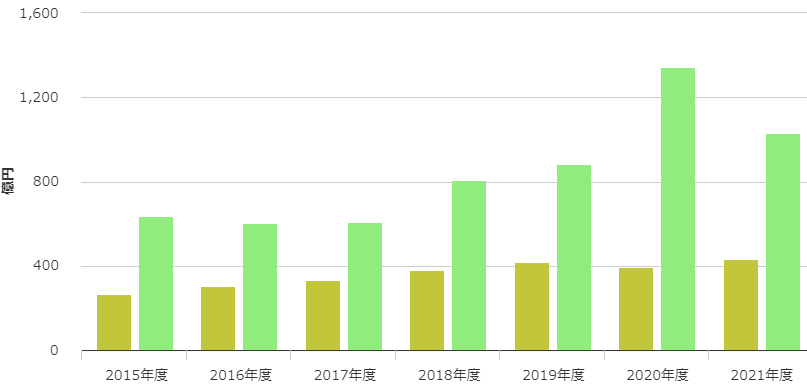
一方、下図に示したアクテムラの国内売上(ウグイス色)をみると、海外(緑色)と同様に年々増加しています。(中外製薬株式会社ホームページより)。
アクテムラが、最初に取得した適応症(希少疾患のキャッスルマン病)だけでビジネスを継続していたら、年間売上数億円の商品に留まっていたかもしれません。
ところが実際は、関節リウマチ、若年性突発性関節炎、成人スチル病、サイトカイン放出症候群などの適応症を追加した結果、2021年には国内外合算して、約1,500億円の商品に成長しています。
まさに、アクテムラの開発は適応症拡大の好事例と言えます。
![]()

コメント