
薬学部の学生さんや製薬企業の若手の皆さんに、医薬品の開発がどのように進められたのかを知っていただくために、医薬品の研究開発経験者の視点で、審査報告書の中から要点を探ってみようと思い立ち、このブログを立ち上げました。ブログの書き方が不慣れですが、これからポリッシュアップしていきますので、どうぞよろしくお願いいたします! [第4回]
今回深掘りするコミナティ筋注は、コロナ禍の2021年2月に下記の効能・効果と用法・用量で特例承認されました。
- 効能・効果:SARS-CoV-2による感染症の予防
- 用法・用量:日局生理食塩液1.8mLにて希釈し、1回0.3mLを合計2回、通常、3週間の間隔で筋肉内に接種する。(1バイアル中にトジナメラン0.225mgを含有しているので、1回の投与量は37.5µgです)
2022年10月17日公表(首相官邸)時点で、日本国内の接種人数は、1回接種104,248,072人、2回接種102,891,831人、3回接種82,755,821人、4回接種37,202,335人となっており、人口の八割以上が2回接種を受けたことになります。
米国CDC(アメリカ疾病予防管理センター)はCOVID-19ワクチンの安全性情報を収集しています。 Safety of COVID-19 Vaccines | CDC 2022年10月24日付のアップデートの中で、「COVID-19ワクチンは安全で有効でワクチン後の重篤な反応は稀」としたうえで、重症型の副反応として「アナフィラキシー」「血栓を伴う血小板減少症候群」「心筋症と心膜炎」「ギラン・バレー症候群」を報告しています。
最近、SNS上でもコロナウイルスワクチンの副作用についての話題が目に付くようになり、本剤の原薬・製剤に関することや、開発(非臨床を含めて)においてどのような安全性情報があがっていたのか気になるところです。
気になる医薬品の審査プロセスを知るために、審査報告書を深掘りしてみましょう!
(文中に「?」と記載しているのは、審査報告書内で黒塗りにされている箇所です)
コロナウイルス修飾ウリジンRNAワクチン(SARS-CoV-2)[コミナティ筋注]の審査報告書はこちら⇒672212000_30300AMX00231_A100_6.pdf (pmda.go.jp)
トジナメラン(有効成分名)は、
- SARS-CoV-2のスパイクタンパク質類縁体(Lys986Pro、Val987Pro)全長をコードするmRNA
- 5’キャップ構造及びポリA配列を含み、全てのウリジン残基がN1-メチルシュードウリジン残基に置換された、4284個のヌクレオチド残基からなる1本鎖RNA:すべてのUTPがm1ΨTPに置換するのは、mRNAに対する免疫原性の抑制及び翻訳の促進のため。
まとめ
今回コミナティ筋注を取り上げたのは、今年(2022年)の後半には5回目のワクチン接種が始まる一方で、SNS上で副作用の話題が散見されるようになったからです。
この審査報告書を読んでの感想は次のとおりです(すべて個人的見解です)。
- 2020年にコロナパンデミックが起きた際には、新型コロナウイルスのワクチンを大至急で用意しなくちゃいけないという、衛生的かつ政治的なプレッシャーの中、mRNAワクチンがスピーディーに開発されたのは、素晴らしい!
- 外来タンパク質であるスパイクタンパク質のmRNAが全身に行きわたり、様々な臓器(肝臓、脾臓、副腎、卵巣など)でスパイクタンパク質が発現し、抗原特異的なTh1免疫反応が惹起され、臓器障害が起こる可能性は否定できない。
- mRNA製剤は画期的な技術で、特定の遺伝子欠損の患者さんに、当該遺伝子mRNAを導入するのであれば良いのだろうけど、外来タンパク質を導入するのは、ちょっと抵抗はある。
- 臨床パートで示されたように、2回接種できちんと有効性が認められたにもかかわらず、実臨床では何故3回以上接種することになったのでしょうか? 治験で確認されたのは2回接種の安全性・有効性であり、3回以上接種の安全性・有効性データはあったのだろうか?
厚生労働省の、厚生科学審議会 (予防接種・ワクチン分科会 副反応検討部会) (mhlw.go.jp) では、定期的にワクチン接種による副反応疑い報告が共有されています。
令和4年1月からの「副反応疑い報告」を以下のとおり添付しました(令和4年以前のデータも厚生労働省のウエブサイトに掲載されています)。関心のある方は是非ご覧になって下さい。冒頭お伝えした「アナフィラキシー」「血栓を伴う血小板減少症候群」「心筋症と心膜炎」「ギラン・バレー症候群」は国内でも確認されています。死亡例が思っていたよりも多いこと、本薬との因果関係の評価が情報不足により出来ない症例(表中の因果関係評価の欄に「γ」で表示)が多すぎることが、少し気になりました(別の機会にデータ集計してみたいです)。
- 000898940.pdf (mhlw.go.jp) 令和4年1月3日から令和4年1月23日報告分まで
- 000914317.pdf (mhlw.go.jp) 令和4年1月24日から令和4年2月20日報告分まで
- 000928673.pdf (mhlw.go.jp) 令和4年2月21日から令和4年3月20日報告分まで
- 000938139.pdf (mhlw.go.jp) 令和4年3月21日から令和4年4月17日報告分まで
- 000948915.pdf (mhlw.go.jp) 令和4年4月18日から令和4年5月15日報告分まで
- 000962318.pdf (mhlw.go.jp) 令和4年5月16日から令和4年6月12日報告分まで
- 000972958.pdf (mhlw.go.jp) 令和4年6月13日から令和4年7月10日報告分まで
- 000983370.pdf (mhlw.go.jp) 令和4年7月11日から令和4年8月7日報告分まで
- 000998687.pdf (mhlw.go.jp) 令和4年8月8日から令和4年9月4日報告分まで
- 001010919.pdf (mhlw.go.jp) 2022年9月5日~2022年10月9日報告分まで
![]()
起原又は発見の経緯及び外国における使用状況に関する資料等
コロナウイルスは、
- ニドウイルス目コロナウイルス科に属する一本鎖ポジティブ鎖RNAウイルスである。
- これまでヒトに日常的に感染し、風邪を引き起こすコロナウイルス(HCoV)として、HCoV-229E、HCoV-OC43、HCoV-NL63、HCoV-HKU1の4種類が知られていた。
- 近年になり動物からヒトに感染し重症肺炎を引き起こすコロナウイルスとして、2003年にSARS-CoV、2012年にMERSCoVが同定されている。
SARS-CoV2は、
- 2019年12月31日、中国湖北省武漢市における原因不明の肺炎の発生をWHOに報告した。
- 2020年1月12日、WHOは当該肺炎が新型コロナウイルスによるものであると発表した。
- 2020年1月30日、WHOは、中国湖北省武漢市における新型コロナウイルス関連肺炎の発生状況が国際的に懸念される公衆衛生上の緊急事態に該当すると発表した。
- 2020年2月11日、新型コロナウイルスをSARS-CoV-2、SARS-CoV-2 による疾患をCOVID-19と命名した。
- 2021年1月17日時点で、世界での総感染者数は93,217,287例、総死亡例は2,014,957例であり、WHO の国・地域分類における感染者数及び死亡者数の、総感染者数及び総死亡者数に対する割合は、アメリカ44%及び47%、ヨーロッパ33%及び33%、東南アジア13%及び9%、東地中海6%及び6%、アフリカ2%及び3%、西太平洋1%及び1%である。
本邦では、
- 2020年1月15日に1例目のSARS-CoV-2に関連した肺炎の患者が確認された。
- 2020年2月1日、新型コロナウイルス感染症が感染症の予防及び感染症の患者に対する医療に関する法律(感染症法)に基づく指定感染症及び検疫法に基づく検疫感染症に指定された。
- 2020年4月7日、改正新型インフルエンザ等対策特別措置法に基づく1度目の緊急事態宣言が行われた。
- 2020年5月25日に緊急事態解除宣言が行われた。新規感染者数(PCR検査陽性者数)は、一旦は減少傾向となったが、同年10月頃から再度増加傾向となった。
- 2021年1月7日に2度目の緊急事態宣言が行われた。
- 2021年1月19日時点、本邦での感染者数は332,231例、死亡者数は4,547例、これに加え、空港検疫で2,082例、チャーター便による海外からの帰国者で15例が確認されており、計334,328例、うち死亡例は空港検疫での死亡1例を加えて4,548例と報告されている。
- 2020年2月3日に横浜港に到着したクルーズ船「ダイアモンド・プリンセス号」の乗客での感染者数は712例、死亡者数は13例と報告されている。
COVID-19は、
- 初期症状は、インフルエンザや感冒に似ており、発症初期に判別することは困難である。
- SARS-CoV-2曝露から発症までの潜伏期間は1~14日間であり、通常は5日程度で発症することが多い。
- 発症前から感染性があり、発症から間もない時期の感染性が高いこと及び無症候性の場合もあることが市中感染の原因とされており、ウイルスの伝染を制御することを困難にしている。
- 発熱、咳嗽、倦怠感、呼吸困難、味覚障害、嗅覚障害等の症状が多くの患者に認められ、約80%の患者は軽症のまま1週間程度で治癒するが、約20%は肺炎症状が増悪し、約5%は人工呼吸器を必要とする急性呼吸窮迫症候群や多臓器不全に至り、2~3%が致命的な経過をたどる。
- 2021年1月20日時点において本邦で「SARS-CoV-2による感染症」の治療に対して承認されている医薬品はレムデシビルがあり、デキサメタゾンは既承認の効能・効果の範囲で使用可能であるが、これらの治療を行っても、本邦の感染者、重症者及び死亡者の報告数は増加が続いており、医療体制のひっ迫も問題となっている。そのため、感染拡大対策として、SARSCoV-2ワクチンによるCOVID-19の発症予防が期待され、早期のワクチン開発が求められている。
本剤の開発は、
- 2021年1月時点で、本邦で承認されている、SARS-CoV-2による感染症の予防等を目的とするワクチン等の医薬品はない。
- 本剤は、SARS-CoV-2のSタンパク質をコードするmRNAを有効成分とするワクチンである。コードされた標的タンパク質を持続的かつ効率的に翻訳するためmRNA の塩基配列が最適化され、また、生体内でのRNA分解を抑制し、mRNAの細胞内へのトランスフェクションを可能とするためmRNAをLNPに封入している。
- 2020年?月より独国BioNTech 社及び米国Pfizer社により本剤の開発が進められた。目的は、SARS-CoV-2による感染症の予防。
- 2020年4月より海外臨床試験(C4591001試験)は同年4月より開始された。
- 2020年10月より国内臨床試験(C4591005試験)は同年10月より開始され、2021年1月20日時点で海外臨床試験(C4591001試験)とともに試験継続中である。
- 2020年12月21日に条件付き承認がなされた。これは、海外C4591001試験におけるCOVID-19発症予防効果及び本剤2回目接種後2カ月時点の安全性のデータに基づき、COVID-19の予防に対して、米国では2020年12月11日にEmergency Use Authorizationを得られたから。
- 2020年12月18日にファイザー株式会社により本邦で製造販売承認申請された。これは、米国FDAによるEmergency Use Authorizationが得られたこと、製造販売承認申請日時点で、欧州では審査中であったこと、本邦では免疫原性及び安全性を評価する国内C4591005試験が実施中であったことから。
本報告書は、「特例承認の検討がなされている医薬品の取扱いについて(依頼)」(令和2年12月17日付け薬生薬審発1217第2号)を踏まえ、申請者から提出された資料に基づき審査された。
品質に関する資料及び機構における審査の概略
「原薬」
- 本薬(BNT162b2)は、SARS-CoV-2(Wuhan-Hu-1株由来)のS(スパイク)タンパク質S1及びS2をコードするmRNAである。
- Sタンパク質の最適な融合前構造を保つため、2アミノ酸が置換(K986P及びV987P)されるよう設計した。
- 5’末端にキャップ構造、翻訳の効率化のための配列(?)、小胞体輸送のためのシグナル配列(?)、RNAの安定化のための配列(?)及びポリA鎖を含み、mRNAに対する免疫原性の抑制及び翻訳の促進のため、すべてのUTP がm1ΨTP(N1-メチルシュードウリジン残基)に置換した。:ここに3種の?で情報が秘匿されています。この審査報告書では本薬の全核酸配列は開示してあるのですが、技術的工夫のある配列は特定してくれません。
- 製造方法:
- 直鎖鋳型DNAを鋳型に、? 、ATP、CTP、m1ΨTP、GTPを用いてin vitro転写反応により製造される。:ここに記載の「?」はおそらく転写酵素でしょうね。
- 製造工程は、in vitro転写反応、? 、? 、?及び?であり、すべて重要工程とした。
- 直鎖鋳型DNAは、WCBを培養後、回収された大腸菌を溶菌して環状プラスミドDNAを得て、環状プラスミドDNAを、及びにより精製後、処理し、調製する。直鎖鋳型DNAの管理項目として、性状(濁度及び色調)、pH、吸光度(?)、制限酵素解析(?の完全性)、純度、残存タンパク量、微生物限度、エンドトキシンを設定した。
- 製造工程の開発の経緯(同等性/同質性):
- Process 1:非臨床試験及び臨床試験で用いられた原薬を製造。原薬は?により作製した鋳型DNAを用いてin vitro転写反応後、?を経て、?により精製された。
- Process 2:市販予定製剤を製造。原薬はプラスミドDNAから作製した直鎖鋳型DNAを用いてin vitro転写反応後、?及び?を経て、?及び?により精製された。
- Process 1からProcess 2への変更に際して、品質の同等性/同質性が確認された。
- 製造工程の開発にはQbDの手法が利用された。
- 特性解析:
- 一次構造:
- RNA配列(RNase T1処理後、オリゴヌクレオチドマッピング(IP-RP-HPLC/ESI/MS/MS又はLC-MS/MS))
- 5’キャップ構造(RNase H処理後、IP-RP-HPLC-UV/ESI MS 及びLC-UV/MS)
- ポリA鎖(RNase T1処理後、IP-RP-HPLC-UV/ESI MS 及びLC-UV/MS)
- 高次構造:高次構造(円偏光二色性スペクトル)
- 一次構造:
- 目的物質由来不純物:二本鎖RNA
- 製造工程由来不純物:
- 鋳型DNA、工程由来不純物A※、工程由来不純物B※、ATP、CTP、GTP、m1ΨTP、工程由来不純物C※、工程由来不純物D※、工程由来不純物E※、酢酸マグネシウム、塩化カルシウム、硫酸アンモニウム、Triton X-100、トリス塩酸緩衝液、グリセロール、塩化ナトリウム、塩化カリウム、工程由来不純物F※、工程由来不純物G※及び工程由来不純物H※とされた。
- 残存鋳型DNAは、原薬の規格及び試験方法により適切に管理されている。工程由来不純物A※及び工程由来不純物B※は、製造工程において十分に除去されることが確認されている。
- ATP、CTP、GTP、m1ΨTP、工程由来不純物C※、工程由来不純物D※、工程由来不純物E※、酢酸マグネシウム、塩化カルシウム、硫酸アンモニウム、Triton X-100、トリス塩酸緩衝液、グリセロール、塩化ナトリウム、塩化カリウム、工程由来不純物F※、工程由来不純物G※及び工程由来不純物H※は、精製工程で不純物を除去できないと仮定しても、本剤の接種量に対して安全性に問題がない量であることが確認されている。
- 原薬の規格及び試験方法:含量規格、性状、確認試験(RT-PCR)、pH、純度試験(二本鎖RNA(免疫ブロット)及び鋳型DNA(qPCR))、5’キャップ(逆相HPLC)、ポリA 鎖(ddPCR)、RNA 完全性(キャピラリーゲル電気泳動)、エンドトキシン、微生物限度、定量法(紫外可視吸光度測定法)が設定された。
- 原薬の安定性
- 長期保存試験:-20±5℃(3カ月又は6カ月にて実施)
- 加速試験:5±3℃(3カ月又は6カ月にて実施)
「製剤」
- 製剤は、希釈した原薬と、LNP(脂質ナノ粒子) を構成する脂質(ALC-0159、ALC-0315、DSPC 及びコレステロール)を混合して製造される。:ALC-0159とは、2- [(polyethylene glycol)-2000]-N, Nditetradecylacetamide、ALC-0315とは、[(4-hydroxybutyl) azanediyl] bis (hexane-6,1-diyl) bis(2-hexyldecanoate)のことで、ALC-0159及びALC-0315はヒトへの使用前例がない。DSPCとは、1,2-distearoyl-sn-glycero-3-phosphocholine。
- 1バイアル(0.45mL)あたり原薬0.225mg を含有する複数回接種用のバイアル製剤である。使用時に、生理食塩液1.8mLにて希釈し、全量2.25mLとする。製剤には、精製白糖、塩化ナトリウム、塩化カリウム、リン酸水素ナトリウム二水和物、リン酸二水素カリウム及び注射用水が添加剤として含まれる。本剤は、1バイアルから5回分採取可能な製剤として申請された。
- 規格及び試験方法:含量規格、性状、確認試験(脂質(HPLC)及びRNA(RT-PCR))、浸透圧、pH、RNA完全性(キャピラリーゲル電気泳動)、封入RNA(蛍光分析)、粒子径及び粒子の多分散性(動的光散乱法)、エンドトキシン、採取容量、不溶性微粒子、無菌、脂質含量(HPLC)、定量法(蛍光分析)が設定された。審査の過程において、生物活性(?)が追加された。:生物活性はスパイクタンパク質のin vitro発現ですかね?
- 製剤の安定性:長期保存試験を-70±10℃(原薬Process 1、6カ月)、-90~-60℃(原薬Process 2、3カ月)で実施した。
「QbD」(Quality by Design)
- CQA(Critical Quality Attribute)の特定
本剤の有効性及び安全性に影響を及ぼす品質特性として、以下のCQAが特定された。- 原薬のCQA:性状(濁度及び色)、pH、RNA完全性、微生物限度、エンドトキシン
- 製剤のCQA:粒子径及び粒子の多分散性、封入RNA、RNA含量、脂質(ALC-0159、ALC-0315、DSPC及びコレステロール)含量、in vitro発現、RNA完全性、5’キャップ構造、ポリA鎖
- 工程の特性解析
工程パラメータに基づき、各工程の特性解析が実施された。 - 管理方法の策定
上記の工程特性解析を含む工程知識、製品品質特性、安定性試験等に基づき、工程パラメータ及び性能特性の管理、並びに規格及び試験法の組合せによる本剤の品質特性の管理が策定された。
「機構における審査の概略」
(1)原薬の製造方法の変更について
申請者の説明は、
- Process 1で製造した原薬4ロットとProcess 2で製造した5ロットについて、性状、pH、確認試験(RTPCR)、含量、RNA完全性(キャピラリー電気泳動)、5’キャップ構造(逆相HPLC)、ポリA 鎖(ddPCR)、鋳型DNA(qPCR)、二本鎖RNA(免疫ブロット)及び浸透圧を比較した結果、大きな差異はなかった。
- Process 1と比較してProcess 2の原薬は、RNA完全性が低い傾向を示したが、いずれも規格に適合していた。:具体的な規格値などの秘密事項は審査報告書には記載されません。
- Process 1で製造した3ロットとProcess 2で製造した1ロットについて、ポリA鎖含量にわずかな差が認められたが、すべてのロットが規格に適合していた。また、質量分析及び分光分析により、一次構造及び高次構造が同等であることが確認された。in vivoでのタンパク質抗原の翻訳に重要と考える5’キャップ構造を有する完全なRNAの割合に大きな差異はなかった。
以上より申請者は、Process 1とProcess 2の原薬に、安全性及び有効性に影響を与えるような差異はなく、同等性/同質性は示されたと回答した。
PMDAは申請者の説明を了承した。
(2)不溶性異物について
製剤のロット分析の「不溶性異物」において、白色~灰白色の不透明な無晶性の粒子が検出されている。PMDAは当該粒子について説明を求めた。
申請者の説明は、
- 「白色~灰白色の不透明な無晶性の粒子」は、? ~? %で認められ、脂質供給元、製造所及び充填工程における発生の相関は認められなかった。
- 経時的な粒子の増減はなく、粒子を解析した結果、製剤に含まれるRNA及び脂質であることが判明した。
- 粒子は生理食塩液で希釈することで消失すること、粒子の有無によりRNA含量及び封入RNAの割合に違いがないことを確認した。
以上から申請者は、本剤中に粒子が生じても、本剤の有効性及び安全性に影響を与えることはないと回答した。:本剤が発売されてから、バイアル溶液中に粒子がある!、とニュースで報じられていましたが、この粒子の事でしたら問題ないようです。
なお、稀に生理食塩液で希釈後の溶液中にも粒子が存在することが確認されているため、添付文書において、生理食塩液で希釈した液に粒子が認められないことを確認した上で使用し、粒子が認められた場合は使用しない旨を記載する予定。
PMDAは、申請者の説明を了承した。
(3)原薬及び製剤の有効期間について
申請者は、本剤の原薬及び製剤の有効期間について、いずれも海外で設定されている有効期間と同じ6カ月と設定している。PMDAは、原薬及び製剤の有効期間について説明を求めた。
申請者の説明は、
- Process 1とProcess 2で製造された原薬の同等性/同質性は示された。原薬の有効期間について、Process 1で製造した原薬2ロットの長期保存試験及び加速試験においてRNA完全性及びRNA 含量に変化は認められなかったため、原薬の有効期間を6カ月と設定することは可能と考える。
- なお、Process 2で製造した原薬4ロットについて長期保存試験を継続中であり、6カ月を超えてデータを取得する予定である。
- 製剤の有効期間について、Process 1の原薬を用いた製剤2ロットの長期保存試験において、6カ月時点までの主な品質特性(RNA完全性、封入RNA、粒子径及び粒子の多分散性、RNA含量等)に大きな変化がないことを確認した。
- また、Process 2 の原薬を用いた製剤2ロットの長期保存試験は、生物活性のデータは得られていないものの、3カ月まですべて規格に適合していることから、製剤の有効期間を6カ月と設定することは可能と考える。なお、Process 2の原薬を用いた製剤2ロットについて長期保存試験を継続中であり、6カ月を超えてデータを取得する予定である。:通常、生物活性データなしで承認は得られません。本剤については、コロナ禍の特例承認です。
この説明に対してPMDAは
- 原薬の有効期間について、Process 1で製造した原薬2ロットの長期保存試験は規格試験項目のうち一部の項目のみ試験が実施されていること、また、ロット数もICH Q5Cで例示されている3ロットに満たないことから、6カ月までの原薬の安定性を確認するにはさらに追加の情報が必要。:通常、3ロットのデータが求められます。
- しかしながら、提出された安定性試験成績において経時的に大きな変化は認められていないこと、また、Process2で製造した原薬の長期保存試験において最初の1ロットがRNA完全性の規格に適合しなかったものの、当該ロットは試験開始時から規格の下限(? %)に近い値を示していたこと、当該ロットの後に製造された連続3ロットの長期保存試験では3カ月時点まですべて適合していることから、現時点で原薬の安定性に大きな問題は認められていないと判断した。
- 製剤の有効期間について、提出された6カ月の長期保存試験成績は2ロットのみであり、6カ月までの製剤の安定性を確認するにはさらに追加の情報が必要と考える。しかしながら、Process 2の原薬を用いた製剤の3カ月時点までの長期保存試験成績も含め、提出された試験成績はすべての規格に適合しており、経時的に大きな変化は認められていない。
- 本剤は各国共通の原薬から製剤が製造され、各国へ供給されている。原薬及び製剤の有効期間を別途設定することは製造管理及び流通管理の上で支障となり、日本に供給されるロットや数量に影響する可能性があることから、現在のCOVID-19流行状況及び本剤の社会的必要性を鑑みると、現時点では原薬及び製剤の有効期間を海外の有効期間と同じ6カ月と設定することはやむを得ないと判断した。
- ただし、現在実施中である原薬及び製剤の長期保存試験の試験成績については、取得後速やかに機構に提出する必要があると考える。
以上を踏まえPMDAは、原薬の有効期間は?製容器を用いて-20±5℃で保存するとき6カ月、製剤の有効期間は-90~-60℃で保存するとき6カ月とする、と回答した。
(4)新添加剤について
製剤には、新添加剤として、使用前例がないALC-0159及びALC-0315並びに「特定の製剤や特定の条件下においてのみ使用が認められた添加物の取扱いについて」(平成21年6月23日付け事務連絡)において特定の製剤又は特定の条件下においてのみ使用が認められているDSPCが含まれる。
申請者は、各添加剤の使用理由について、ALC-0159は本剤と血漿タンパク質との相互作用を抑えること、ALC-0315は本剤の粒子形成、細胞への取込み及び本剤に含有されるRNAのエンドソームからの放出を調節すること、DSPC は「?」することを目的としている旨を説明している。:DSPCはALC-0159とALC-0315から成る脂質ナノ粒子の構造を安定化させることを目的に使用していると考えられます。
- 規格及び試験方法並びに安定性について
- PMDAは、ALC-0159、ALC-0315及びDSPCの規格及び試験方法並びに安定性について、提出された資料から問題はないと判断した。
- 安全性について
- 申請者は、ALC-0159、ALC-0315及びDSPCの単回投与毒性、反復投与毒性及び生殖発生毒性について、本剤の毒性試験の結果に基づき説明した。
- これら新添加剤の遺伝毒性について、投与経路の異なる使用前例、構造活性相関による変異原性評価(専門的経験に基づくルールベースの方法及び統計ベースの方法)等により安全性に懸念がないと説明している。
以上の申請者の説明に対してPMDAは、
- ラットにおける反復筋肉内投与毒性試験では、肝臓への影響(血中GGTの増加及び肝細胞の空胞化)が認められているが、毒性学的意義は低いと考えられる。
- ALC-0159、ALC-0315及びDSPCは本剤の製剤特性を担保するために必要と考えられることから、本剤にこれらの添加剤を使用することは可能と考える。
- しかしながら、本剤の毒性試験では長期間の反復投与毒性が評価されていない。:当初、新型コロナワクチンは2回投与としていましたが、2022年には4回目投与が始まっており、現時点で長期間の反復投与毒性の評価を求めても良いと思います。
以上よりPMDAは、これらの添加剤は、本剤の用法・用量に限った使用とすべきであり、使用前例として取り扱わないことが適切と判断した。:今年(2022年)9月に、ファイザー社のオミクロン株2価ワクチンが承認されましたが、これらの添加剤を使用する場合は長期間の反復毒性試験を求められたのですかね?
非臨床薬理試験に関する資料及び機構における審査の概略
「効力を裏付ける試験」
(1)in vitroにおける抗原発現
- トランスフェクション試薬と混合した本薬(BNT162b2)をHEK293T細胞内に導入し、抗原(SARSCoV-2由来全長Sタンパク質)の発現がウエスタンブロット法及び免疫蛍光法で評価した結果、
- HEK293T細胞において抗原の発現が確認され、発現した抗原は小胞体に局在していたことから、抗原が小胞体で合成され、プロセシングを受けることが示唆された。:ここに記載のin vitro試験で用いた本薬(BNT162b2)のRNA量が記載されていませんね。スパイクタンパク質のin vitroでの発現量について用量依存性は確認したのかな?
- 本剤(LNP封入BNT162b2)又はトランスフェクション試薬と混合した本薬(BNT162b2)をHEK293T細胞内に導入し、細胞への導入効率及び細胞の生存率がフローサイトメトリーで評価した結果、
- 本剤群及び本薬群における抗原発現細胞はそれぞれ98.0±0.2%及び85.1±4.4%であった。
- 本剤群及び本薬群の細胞の生存率は対照群(非トランスフェクション処理細胞)と同程度であった。
- Expi293F細胞に、本薬と同じアミノ酸配列をコードするDNAによりSタンパク質を発現させたところ、発現したSタンパク質のヒト細胞受容体であるアンジオテンシン変換酵素2及びヒト抗RBD中和抗体への結合、並びに細胞表面への発現が確認された。:RBDとは受容体結合ドメインのこと。
(2)マウス免疫原性試験
- BALB/c マウス(雌8例/群)に本剤(RNA量として0.2、1.0 又は5.0μg)を単回筋肉内投与した際の免疫応答が評価した結果、
- Sタンパク質S1及びRBDに対する特異的IgG抗体の検討(ELISA法):検討された測定時点(本剤投与7~28日後)の血清中S1特異的IgG抗体及びRBD特異的IgG抗体を測定した結果、用量依存的な抗原特異的抗体の産生が認められた。
- シュードウイルスを用いた中和抗体の検討(中和法)検討された測定時点(本剤投与14~28日後)の血清中の中和抗体をシュードウイルスを用いて測定した結果、用量依存的な中和抗体の産生が認められた。
- IgGサブタイプの検討(ELISA法):本剤投与28日後の血清中IgGサブタイプ(IgG1及びIgG2a)について検討した結果、Th2細胞優位の免疫応答は認められず。
- 脾臓細胞におけるサイトカイン産生の検討(Luminex法及び細胞内サイトカイン染色法):本剤投与28日後の脾臓細胞をS1又はRBDペプチドにより刺激した結果、IFN-γ、TNF-α、IL-2、IL-6、IL-18及びGM-CSFの産生が認められた一方で、IL-4、IL-5及びIL-13の産生はわずかであり、Th1細胞優位の免疫応答が示唆された。
- 対照群(緩衝液投与28日目の脾臓細胞)と比較して、本剤投与群におけるIFN-γ、TNF-α 又はIL-2を産生するCD4又はCD8陽性T細胞の割合は増加したが、IL-4を産生するCD4陽性T細胞の割合に有意な増加は認められなかった(IL-4はCD4陽性T細胞のみで評価された)。:これもTh1細胞優位の免疫反応です。
(3)サル攻撃試験
アカゲザル(雄6例/群)に、本剤を21日間隔で2回筋肉内投与したときの免疫応答、及びSARS-CoV-2 曝露後の感染防御/発症予防効果を評価した。
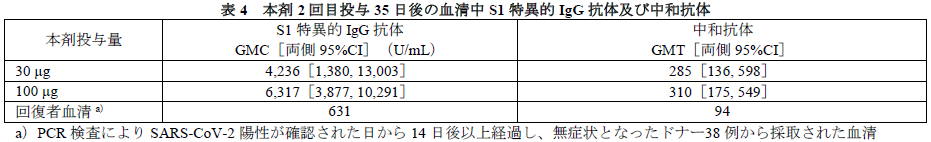
- S1特異的IgG抗体及び中和抗体の検討(ELISA 法及び中和法):本剤(RNA量として30又は100μg)2回目投与35日後における血清中S1特異的IgG抗体及び中和抗体を測定した結果、下表のとおり、いずれの抗体産生も認められた。
- 末梢血単核球におけるサイトカイン産生の検討(ELISpot法及び細胞内サイトカイン染色法):本剤(RNA量として30又は100μg)を投与したサルから採取した末梢血単核球をSタンパク質ペプチドにより刺激し、サイトカイン産生量(IFN-γ及びIL-4)及びサイトカイン産生細胞数が評価された。IFN-γは、本剤2回目投与7日以降の末梢血単核球において高値であったが、IL-4はいずれの時点でも低値であった。また、本剤1回目投与14日以降にIFN-γ、IL-2又はTNF-αを産生するCD4陽性T細胞数及びIFN-γを産生するCD8陽性T細胞数の増加が認められたが、IL-4を産生するCD4陽性T細胞数の増加はわずかであった。以上の結果から、申請者は、本剤を投与したサルにおいてTh1細胞優位の免疫応答が誘導されたと説明している。
- SARS-CoV-2曝露後の感染防御/発症予防効果の検討:本剤(RNA量として100μg)又は生理食塩液を2回目投与55日後のサル(本剤群6例、対照群3例)の鼻腔及び気管内に、SARS-CoV-2(USA-WA1/2020株、1.05×10^6 PFU)を曝露させた後の観察及び検査結果は下表のとおりであった。申請者は、本剤群では対照群に比べて気道から検出されたウイルスRNAは低値であったことから、本剤投与による感染防御効果が示されたと説明している。:スワブとは綿棒採取のこと。鼻腔や中咽頭のスワブでは対照群でも日を追って検出例が減るんですね。

「安全性薬理試験」
本剤を用いた安全性薬理試験は実施されていないが、本剤の安全性薬理は、ラット反復筋肉内投与毒性試験における一般状態観察等から評価され、申請者は、本剤投与による心血管系、呼吸器系、中枢神経系等の生理機能への影響は認められていないと説明した。:通常、安全性薬理試験の実施は求められますが、コロナ禍の緊急対応で省略されたのでしょうね。外来蛋白であるスパイクタンパク質が体内の各臓器で高発現した場合の安全性はどうなのか知りたいですね。
「機構における審査の概略」
本剤の作用機序について
申請者の説明は、
- 本剤のin vitroの検討で、産生されたSタンパク質の宿主細胞での発現、マウス及びサルでの検討で、中和抗体の産生、Th1細胞優位の免疫応答及びIFN-γを産生するCD8陽性T細胞の増加、並びにサルでの検討で、SARS-CoV-2曝露に対する一定の感染防御効果が確認された。:非臨床薬物動態試験で、本剤の一部は全身に分布されたのですが、本剤を取り込んでスパイクタンパク質を発現した細胞がCD8陽性T細胞によって破壊されますね。
- 本剤はSARS-CoV-2の全長Sタンパク質をコードするmRNAを有効成分として含有しており、これにより中和抗体の標的であるSタンパク質が宿主細胞内で産生される。産生されたSタンパク質により液性免疫及び細胞性免疫が惹起されることで、SARS-CoV-2による感染症の予防効果が期待できると考えられる。
この説明に対してPMDAは、申請者の説明を了承した。
(2)変異株に対する中和作用について
本剤の開発が開始されてから現在までに様々なSARS-CoV-2変異株が報告されていることを踏まえ、PMDAは、これらの変異株に対する本剤の中和作用について説明を求めた。
申請者の説明は、
- COVID-19の流行は、当初SARS-CoV-2のSタンパク質がD614(614番目のアミノ酸がアスパラギン酸)であるウイルスが主流であり、本剤に用いられているmRNA配列もD614をコードする。一方、2020年2月頃よりD614G(614 番目のアスパラギン酸がグリシンに置換)であるウイルスが増加し、同年11月時点においてD614Gが世界的に最も累積頻度(89.0%)の高い変異とされている。また、同年12月には、Sタンパク質に複数変異を有し、感染性がより高いとされる変異株が、英国及び南アフリカで報告されている。
- 本剤の被接種者より得られた血清の変異株に対する中和作用を確認するために、Wuhan-Hu-1株のSタンパク質(本剤のmRNAがコードするSARS-CoV-2のSタンパク質と同じ配列を有する)にアミノ酸変異を加えた19種類のSタンパク質遺伝子を用いてシュードウイルスを作製し、本剤の被接種者より得られた血清を用いて各シュードウイルスに対する中和活性を測定した。その結果、検討したすべてのシュードウイルスに対する中和作用が確認された。
- 英国及び南アフリカで報告された変異株に共通するN501Y のアミノ酸変異を有するS タンパク質遺伝子、K417N、E484K 及びN501Y の複数の変異を同時に持つS タンパク質遺伝子、英国で報告された変異株と同じ変異を有するS タンパク質遺伝子を挿入したシュードウイルスに対しても、本剤の被接種者より得られた血清で一定の中和作用が確認されたことから、本剤は種々のSタンパク質変異ウイルスに対して一定の有効性を示すことが期待できると考える。
- ただし、現在流行しているSARS-CoV-2の一部には、中和作用を有するモノクローナル抗体の反応性が低い変異が報告されていること、今後も新たな変異株が出現する可能性があることも考慮すると、変異株に対する本剤の評価は現時点で網羅されているわけではなく、変異株に対する本剤の中和作用については、製造販売後に引き続き情報収集する予定である。:変異株(オミクロン株)に対応すべく、今年(2022年)9月に、オミクロン株2価ワクチン(ファイザー)が承認されました。10月31日にはモデルナ社製の同ワクチンが承認されました。
この説明に対してPMDAは、
- 様々なシュードウイルスに対して、本剤の被接種者より得られた血清の中和作用が確認されていること等の研究結果を踏まえると、2021年1月27日時点で流行している種々の変異株に対して本剤の一定の有効性は期待できると考える。
- しかしながら、SARS-CoV-2のRNA ウイルスとしての生物学的特性や、COVID-19回復者血清の反応性が低い変異株が報告されていること等も考慮すると、今後の流行において、本剤による免疫応答を回避する変異株が出現する可能性もあり、各変異株に対する本剤の中和作用については、製造販売後も引き続き情報収集し、新たな知見が得られた場合には必要に応じて情報提供する等、適切に対応する必要がある。
(3)疾患増強リスクについて
PMDAは、本剤接種による免疫応答により、SARS-CoV-2感染時の症状がワクチン非接種時よりも増強するリスク(疾患増強リスク)について、申請者に説明を求めた。
申請者の説明は、
- SARS-CoV-2ワクチン接種による疾患増強リスクの有無は現時点で不明であるが、SARS-CoV-2に類似するSARS-CoVでは動物試験においてワクチン接種による疾患増強リスクが論文報告されており、Th2型細胞優位の免疫応答が関連することが示唆されている。
- したがって、SARSCoVワクチンと同様のTh2型細胞優位の免疫応答による疾患増強を想定すると、SARS-CoV-2ワクチン接種によりTh1型細胞優位の免疫応答が惹起されれば、SARS-CoV-2感染時の疾患増強リスクは低くなると考えられている。:SARS-CoV-2特有のTh1型細胞優位の免疫応答による疾患増強のリスクは無いのだろうか?
- 本剤の免疫応答を評価した非臨床薬理試験では、本剤投与によりマウス及びサルでTh1型細胞優位の免疫応答が惹起された。また、サル攻撃試験では、本剤接種後SARS-CoV-2を曝露しても、気管支、肺胞、鼻腔及び中咽頭においてウイルスRNA量の速やかな低下が認められ、本剤非接種の対照群と比較して、胸部X線及びCT画像診断による肺の異常所見の程度は軽度であった。さらに、肺の病理組織学的検査でもTh2型細胞優位の免疫応答を示唆する好酸球浸潤を伴う炎症性所見の増悪は認められなかった。
- 加えて、ヒトでの本剤接種によるサイトカイン産生の評価を行った。海外第Ⅰ相試験(BNT162-01試験)では、本剤接種後の被験者から得られた末梢血単核細胞をSタンパク質のペプチドで刺激したところ、CD8陽性T細胞ではINF-γの増加が、またCD4陽性T細胞ではINF-γ及びIL-2の増加が確認されたが、IL-4の増加はほとんど確認されず、Th1型細胞優位の免疫応答が認められた。
以上を踏まえると、申請者は、本剤接種による疾患増強リスクは低いと回答した。:過剰なTh1型免疫応答で、自己の細胞を障害するリスクの評価は?
PMDAは、薬理の観点からの申請者の説明を了承した。ヒトでの疾患増強リスクについては後の項目で引き続き検討する。
非臨床薬物動態試験に関する資料及び機構における審査の概略
本剤又は本薬を用いた非臨床薬物動態試験は実施されていない。
本剤に含有されるLNP又はその構成脂質であるALC-0159及びALC-0315を用いた非臨床薬物動態試験として、吸収、分布、代謝及び排泄に関する試験の成績が提出された。
各項目の測定は次のとおり
- ラットの血漿、肝臓、尿及び糞中におけるALC-0159及びALC-0315の濃度:LC-MS/MS法
- マウスの生体内におけるルシフェラーゼ遺伝子発現量:in vivoイメージングシステム
- LNPを3Hで標識したルシフェラーゼ遺伝子発現mRNA-LNPをラットに投与したときの生体試料中の放射能濃度:液体シンチレーション計数法
- (in vivoイメージングシステム及び液体シンチレーション計数法の定量下限は評価していない)
「吸収」
ルシフェラーゼ遺伝子発現mRNA-LNPの単回静脈内投与試験
ラット(雄3例/時点)にルシフェラーゼ遺伝子発現mRNA-LNPがRNA量として1mg/kg(ALC-0159 1.96mg/kg及びALC-0315 15.3mg/kg)単回静脈内投与され、ALC-0159及びALC-0315の血漿中PKパラメータ及び肝臓中への分布が検討された結果、
- 血漿中のALC-0159及びALC-0315濃度の半減期は分布相ではそれぞれ1.7及び1.6時間、消失相ではそれぞれ72.7及び139時間であった。
- それぞれの血漿中濃度は投与24時間までに最高血漿中濃度の1%未満となった。
- 投与24時間までに速やかな肝臓への分布が認められ、それぞれ投与量の約20%及び約60%が肝臓に分布していると推定された。
「分布」
ルシフェラーゼ遺伝子発現mRNA-LNPの生体内分布
マウス(雌3例/群)にルシフェラーゼ遺伝子発現mRNA-LNPがRNA量として2μg単回筋肉内投与され、in vivoイメージングシステムを利用して投与9日後までの生物発光を測定した結果、
- 最初の測定ポイントである投与6時間後の投与部位及び肝臓領域の発光シグナルはそれぞれ約1.0×10^9及び約5.0×10^7p/sであり、その後は経時的に減少した。
- 発光シグナルは肝臓領域では投与48時間後には検出されず、投与部位では投与9日後に対照群(リン酸緩衝生理食塩液投与群)で検出されたバックグラウンド値付近まで低下した。:発光シグナルが検出されなくなるというのは、ルシフェラーゼタンパク質が分解されたのか、あるいはルシフェラーゼタンパク質を発現する細胞が破壊されたのどちらかで、安全性の観点で意味合いが大きく異なるのではないだろうか?
3H標識ルシフェラーゼ遺伝子発現mRNA-LNPの分布
ラット(雌雄各3例/群)にルシフェラーゼ遺伝子発現mRNA-3H標識LNPがRNA量として50μg単回筋肉内投与され、投与48時間後までの放射能の組織分布を検討した結果、
- 投与部位の放射能濃度は、投与1時間後に最高値(394μg lipid eq./g)を示した後、経時的に減少し、投与48時間後では165μg lipid eq./g であった。
- 投与部位以外で放射能が認められた主な組織は、肝臓、脾臓、副腎及び卵巣であり、投与8~48時間後に最高値(それぞれ26、23、18 及び12μg lipid eq./g)を示した。:スパイクタンパク質が発現するのは投与部位に限局されるのが理想ですが、卵巣などで発現するのはどうなんでしょうか?
「代謝」
ALC-0159及びALC-0315の代謝
- マウス、ラット、サル及びヒトの肝ミクロソーム、S9画分及び肝細胞に、ALC-0159又はALC-0315(最終濃度:肝ミクロソーム及びS9画分では1.5μmol/L、肝細胞では1.0μmol/L)をそれぞれ添加し、37℃で2時間(肝細胞では4時間)インキュベートした後のALC-0159及びALC-0315の未変化体の残存割合は、すべての試料で90%以上であった。
- マウス、ラット、サル及びヒトのS9画分、肝細胞及び血液に、ALC-0159又はALC-0315(最終濃度10μmol/L)をそれぞれ添加し、37℃で24時間(肝細胞では4時間)インキュベートした後の代謝物が検討された。各動物種のS9画分及び肝細胞、並びにマウス及びラットの血液において、ALC-0159ではアミド基の加水分解物、ALC-0315ではエステル基の加水分解物が確認された。
- また、ラットにルシフェラーゼ遺伝子発現mRNA-LNPを単回静脈内投与したときの投与14日までの血漿、尿、糞及び肝臓サンプルを用いて、代謝物が超高速液体クロマトグラフィー質量分析計で測定した結果、ALC-0159の代謝物はいずれの試料からも検出されず、ALC-0315の代謝物はグルクロン酸抱合体が尿中から、及びエステル基の加水分解物がすべての試料中から検出された。
以上の結果から、ALC-0159及びALC-0315はエステル基又はアミド基の加水分解によって緩やかに代謝されることが示唆された。
「排泄」
ALC-0159及びALC-0315の尿糞中排泄
ラット(雄3例)にルシフェラーゼ遺伝子発現mRNA-LNPを単回静脈内投与したときの糞中及び尿中のALC-0159及びALC-0315を検討した結果、
- 投与後336時間(14日)までにALC-0159及びALC-0315の未変化体は糞中にそれぞれ約47.2%及び約1.1%が排泄され、尿中の未変化体はいずれも定量下限値未満であった。
「機構における審査の概略」
(1)本剤の非臨床薬物動態について
PMDAは、本剤を用いた非臨床薬物動態試験は実施されていないことから、本剤の薬物動態について申請者に説明を求めた。
申請者の説明は、
- 本剤はmRNAである本薬をLNPに封入した製剤である。通常、mRNAは生体内に投与されると、生
体内の核酸と同様に速やかに代謝されるが、LNPに封入することでmRNAが代謝されることなく宿主細胞内に取り込まれ、細胞質内でタンパク質を発現することが可能となる。そのためLNPに封入したmRNA製剤の体内動態は、封入されているmRNAによる影響を受けることなく、LNPに依存すると考えられる。 - ルシフェラーゼ遺伝子発現mRNA-LNPを筋肉内投与したときの生体内分布を評価した試験の結果から、本剤を筋肉内投与した場合、本剤は主に投与部位に分布し、一部は全身(主に肝臓)へ一時的に分布し、それぞれでタンパク質を発現するが、いずれの部位でも時間の経過とともに本剤及び発現したタンパク質は消失すると推察された。:ここでは、タンパク質の発現がどの程度の時間持続するかをルシフェラーゼによって評価していますが、その評価は、発現したスパイクタンパク質がルシフェラーゼと同様の消失パターンをたどることを前提にする時に可能だと思います。
機構は、申請者の説明を了承し、提出された非臨床薬物動態試験成績から、本剤の薬物動態特性について一定の把握は可能と判断した。
毒性試験に関する資料及び機構における審査の概略
「単回投与毒性試験」
本剤を用いた単回投与毒性試験は実施されていないが、本剤の単回投与時の毒性(急性毒性)は、ラットにおける反復筋肉内投与毒性試験の初回投与後の結果から評価され、本剤投与による死亡はなく、本剤投与部位における浮腫、体温上昇(雄:+0.54℃、雌:+0.42℃)、白血球の増加、急性期タンパク質の増加等が認められた。
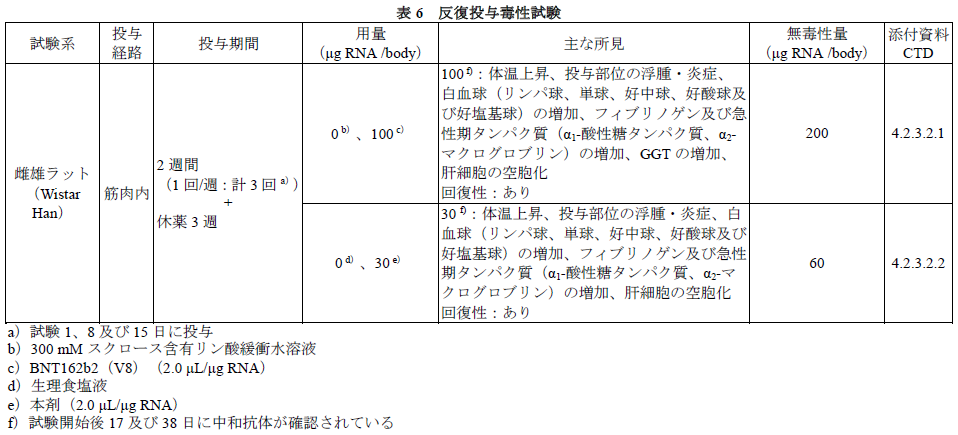
「反復投与毒性試験」
本剤及び本剤のコドン最適化前の製剤(BNT162b2(V8))を用いて、ラットにおける反復筋肉内投与毒性試験が実施された。主な所見は、投与部位における炎症性変化であった。:各臓器の病理組織の結
「遺伝毒性試験」
本剤に含まれるmRNAは天然型の核酸から構成され、新添加剤(ALC-0159、ALC-0315 及びDSPC)にも遺伝毒性の懸念がないことから、本剤を用いた遺伝毒性試験は実施されていない。
「がん原性試験」
本剤は臨床での使用が6カ月以上継続される医薬品ではないことから、本剤を用いたがん原性試験は実施されていない。:当初、新型コロナワクチンは2回投与としていましたが、2022年には4回目投与が実施されている状況から考えると、がん原性試験は追加実施を求めても良いかもしれません。
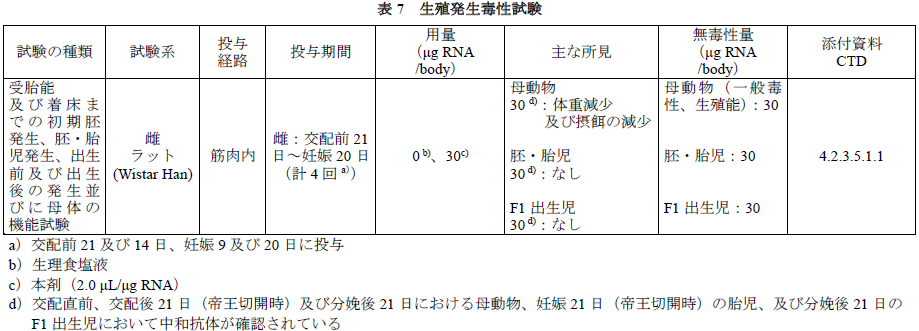
「生殖発生毒性試験」
ラットにおける生殖発生毒性試験が実施された。本剤投与により、親動物及び次世代への影響は認められなかった。
「局所刺激性試験」
本剤の局所刺激性は、ラットにおける反復筋肉内投与毒性試験の結果から評価され、本剤投与部位に回復性のある軽度から中等度の炎症が認められている。
「機構における審査の概略」
(1)肝臓への影響について
PMDAは、ラットにおける反復筋肉内投与毒性試験で認められた血中GGT(γ-GT)の増加及び肝細胞の空胞化について、本剤接種によるヒトでの安全性を説明するよう求めた。
申請者の説明は、
- ラットにおける反復筋肉内投与毒性試験で認められた血中GGTの増加及び肝細胞の空胞化の発現機序は不明である。
- しかしながら、肝細胞の空胞化については、形態学的に脂肪滴に類似し、門脈域の肝細胞に局在すること、本剤に含まれるLNPを用いたラットにおける非臨床薬物動態試験で、脂質の肝臓への分布が確認されていることから、脂質が肝細胞に取り込まれたことにより生じたものと推察される。
- 血中GGTの増加及び肝細胞の空胞化は、いずれも軽度かつ回復性が認められたこと、本剤投与により肝臓及び胆道系への傷害を示唆する病理組織学的所見及び臨床検査値(血中ALT、AST、アルカリホスファターゼ及び総ビリルビン)の変化も認められないことから、いずれも毒性学的意義が低い所見と考える。
- 本剤接種によるヒトでの安全性について、海外C4591001試験の第Ⅱ/Ⅲ相パートにおける肝胆道系の有害事象の発現割合を下表にまとめた。
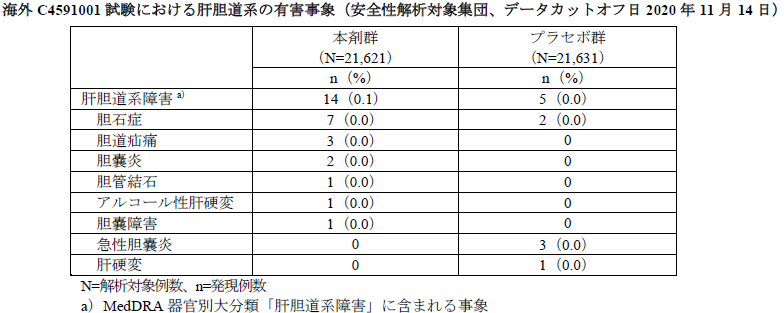
- また、国内C4591005試験において、肝胆道系障害に関する有害事象はデータカットオフ日(2021年1月5日)時点で報告されていない。
以上を踏まえ、申請者は、本剤接種によるヒトでの肝毒性に関するリスクは低いと回答した。:2022年5月末時点で本剤の4回目投与が開始されています。この審査報告書に記載のデータは2回投与後のものなので、市販後調査の結果をみて評価する必要があるでしょうね。
PMDAは、ラットにおける反復筋肉内投与毒性試験で認められた肝臓への影響は、いずれも毒性学的意義が低い所見と判断し、本剤接種によるヒトでの肝毒性に関するリスクは低いとする申請者の見解は受入れ可能、と回答した。
生物薬剤学試験及び関連する分析法、臨床薬理試験に関する資料並びに機構における審査の概略
該当する試験は実施されていない。
臨床的有効性及び臨床的安全性に関する資料並びに機構における審査の概略
「国内第Ⅰ/Ⅱ相試験」(C4591005試験、実施期間2020年10月~継続中:データカットオフ日2021年1月5日)
- 目的:本剤の安全性、忍容性、免疫原性を検討する。:免疫原性とは、抗原が抗体の産生や細胞性免疫を誘導する性質のこと。
- 対象:20~85歳健康人(目標例数160例:本剤群120例、プラセボ群40例、国内2施設)
- デザイン:無作為化観察者盲検のプラセボ対照並行群間比較試験
- 登録例数
- 本剤群:120例
- プラセボ群:40例
- 用法・用量:本剤30μg又はプラセボを21日間隔で2回筋肉内接種
- 結果
- 無作為化された160例(本剤群119例、プラセボ群41例)全例に1回以上治験薬が接種され、全例が安全性解析対象集団とされた。
- 無作為化された160例全例に、1回以上治験薬が接種され、免疫原性測定結果が得られ、全評価免疫原性集団とされた。
- 主要な免疫原性解析対象集団は、評価可能免疫原性集団(事前に規定した期間内に2回目の接種を受け、2回目接種後の免疫原性測定結果が得られた、治験実施計画書からの重大な逸脱がない適格性が確認された集団)と規定されたが、当該集団の結果について、2021年1月29日時点で解析中であり、提出されていない。本報告書では既に得られている全評価免疫原性集団の結果を示す。:つまり、この審査報告書では治験薬を1回以上の受けた人の免疫原性を解析した結果が記載されています。
- 全評価免疫原性集団における治験薬2回目接種後1カ月のSARS-CoV-2血清中和抗体価のGMT及び1回目接種前に対する2回目接種後1カ月のGMFR[両側95%CI]は、本剤群で489.9[420.4, 570.9]及び48.1[41.3, 56.0]、プラセボ群で10.6[9.8, 11.4]及び1.1[1.0, 1.1]であった。:GMTとは幾何平均抗体価、GMFRとは幾何平均上昇倍率のこと。
- 安全性について、観察期間は以下のとおりとされた。有害事象の重症度は予防ワクチンの臨床試験における毒性評価尺度に関するFDAガイダンスに基づき評価された。
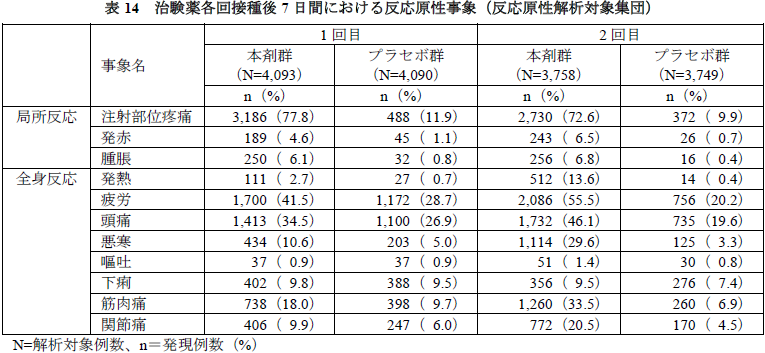
- 反応原性事象(局所反応)(注射部位疼痛、発赤及び腫脹)及び全身反応(発熱、疲労、頭痛、悪寒、嘔吐、下痢、筋肉痛及び関節痛)):治験薬各回接種後7日間(被験者日誌により収集)
- 有害事象(各回接種7日目までに被験者日誌で収集される反応原性事象除く):治験薬1回目接種時から最終接種後1カ月まで
- 重篤な有害事象:治験薬1回目接種時から最終接種後12カ月まで
- 各回接種後7日間に発現した反応原性事象を下表に示す。
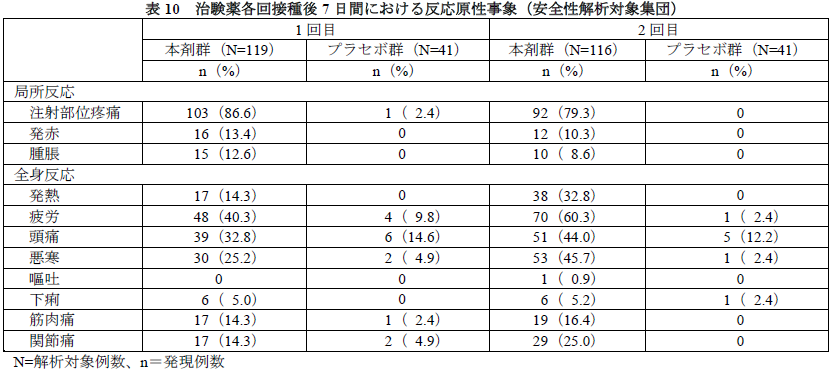
- 有害事象及び副反応(治験薬との因果関係が否定されない有害事象、以下同様)は本剤群10.1%(12/119例)及び1.7%(2/119例)、プラセボ群7.3%(3/41例)及び0例であり、2例以上に認められた有害事象は上咽頭炎(本剤群3例、プラセボ群1例)及び頭痛(本剤群2例、プラセボ群1例)であった。:CDCが報告した重篤な副作用「アナフィラキシー」は、投与直後から30分以内で発症することが多いとされますが、サンプルサイズの小さなこの治験(N=116)では観察されませんでした。
- SARSCoV-2 感染やCOVID-19発症に関連する有害事象は報告されていない。データカットオフ日(2021年1月5日)までに死亡及び重篤な有害事象は認められなかった。
- 中止に至った有害事象は、本剤群1例(悪寒、疲労、注射部位疼痛、関節痛及び頭痛)に認められ、注射部位疼痛及び頭痛は重度であった。いずれも治験薬との因果関係ありと判断され、転帰は回復であった。
「海外第Ⅰ/Ⅱ/Ⅲ相試験」(C4591001試験、実施期間 第Ⅰ相パート:2020年4月~継続中(データカットオフ日 本剤30μg群:2020年11月14日、その他の接種群:2020年8月24日)、第Ⅱ/Ⅲ相パート:2020年7月~継続中(データカットオフ日2020年11月14日)
(1)第Ⅰ相パート
- 目的:本剤の安全性、忍容性及び免疫原性を検討する。
- 対象:18歳以上55歳以下及び65歳以上85歳以下の健康人(目標例数195例:本剤群156例、プラセボ群39例、米国4施設)
- デザイン:無作為化観察者盲検プラセボ対照並行群間比較試験
- 用法・用量:治験薬(本剤10、20、30μg若しくはBNT162b1 10、20、30、100μg、又はプラセボのいずれか)を21日間隔で2回(Day1及びDay22(許容期間はDay19~23))、筋肉内接種。
- 被験者は、各グループ(本剤又はBNT162b1 の用量別及びプラセボと年齢層別の組合せ(BNT162b1 100μgは18歳以上55歳以下のみ)、計13グループ)ごとに、無作為化された。なお、本剤はSARS-CoV-2 のSタンパク質の全長体をコードするmRNAであり、BNT162b1はSARS-CoV-2のSタンパク質のRBDをコードするmRNAである。
- 無作為化された195例(各グループ15例:本剤又はBNT162b1群12例、プラセボ群3例)全例に1回以上治験薬が接種され、全例が安全性解析対象集団とされた。
安全性について、観察期間は以下のとおりとされた。有害事象の重症度は予防ワクチンの臨床試験における毒性評価尺度に関するFDAガイダンスに基づき評価された。
- 反応原性事象(局所反応(注射部位疼痛、発赤及び腫脹)及び全身反応(発熱、疲労、頭痛、悪寒、嘔吐、下痢、筋肉痛及び関節痛)):治験薬各回接種後7日間(被験者日誌により収集)
- 有害事象(治験薬各回接種7日目までに被験者日誌で収集される反応原性事象除く):治験薬1回目接種時から最終接種後1カ月まで
- 重篤な有害事象:治験薬1回目接種時から最終接種後6カ月まで
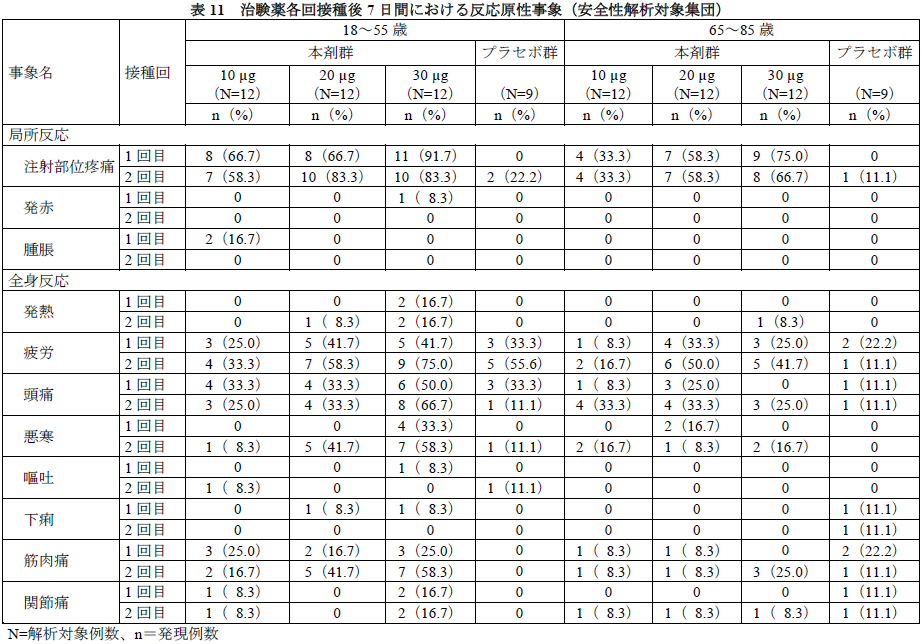
治験薬各回接種後7日間に認められた反応原性事象は下表のとおりであった。
有害事象及び副反応の発現割合は、下表のとおりであった。
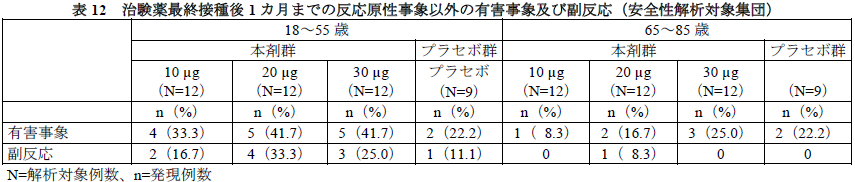
データカットオフ日(本剤30μg 群:2020年11月14日、その他の接種群:2020年8月24日)までに死亡例及び治験中止に至った有害事象は認められなかった。
重篤な有害事象は、本剤30μg群1例に末梢神経損傷(当初は神経炎として報告)が報告され、治験薬との因果関係は否定され、転帰は2020年12月16日時点で未回復であった。
臨床検査値異常について、Grade 3以上の異常変動として、リンパ球数減少が本剤10μg群及び30μg群各1例、ビリルビン増加が本剤10μg群1例に認められた。いずれも1回目接種後1~3日に発現し、1回目接種後6~8日までに基準値範囲内に回復した。
申請者は、本剤接種直後に認められたリンパ球数減少は一過性(接種6~8日で回復)であったことから、リンパ球枯渇ではなくリンパ球再分布によるものと考えられ、臨床的意義は低いと判断して、第Ⅱ/Ⅲ相パートでは臨床検査値は検討しないこととしたと説明している。
(2)第Ⅱ/Ⅲ相パート
安全性について、データカットオフ日(2020年11月14日)までに無作為化された43,548例のうち、43,449例に1回以上治験薬が接種され、このうち同意を取得していなかった本剤群1例を除く43,448例(本剤群21,720例、プラセボ群21,728例)が安全性解析対象集団とされた。また、安全性解析対象集団のうち、被験者日誌が収集された8,183例(本剤群4,093例、プラセボ群4,090例)が反応原性解析対象集団とされた。
有効性について、データカットオフ日(2020年11月14日)までに無作為化された43,651例のうち、3,374例(治験薬未接種又は規定された期間内に2回目が接種されなかった3,111例、2回目接種7日目までに重大な治験実施計画書の逸脱があった371例、無作為化後に適格性基準に合致しないことが判明した62例、同意を取得していなかった1例(重複含む))を除く40,277例(本剤群20,033例、プラセボ群20,244例)が有効性評価可能集団とされ、主要な解析対象集団とされた。
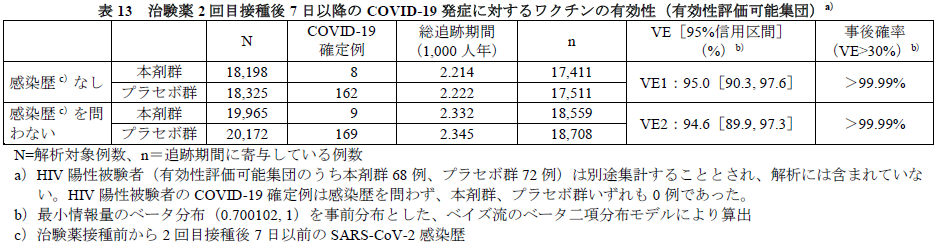
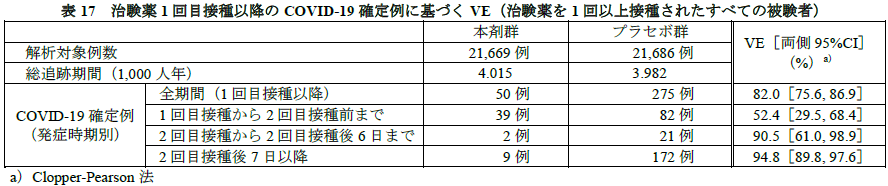
有効性について、主要評価項目は、以下の被験者におけるCOVID-19確定例(1,000人年あたりの治験薬2回目接種後7日以降のCOVID-19発症)に基づくVE(VE1及びVE2)とされた(VE(%)=100×(1−被験者の追跡期間における1,000人年あたりのCOVID-19発症率の本剤群とプラセボ群の比(IRR)))。:VE(vaccine efficacy)とはワクチン効果のこと。
- VE1:治験薬接種前から2回目接種後7日以前にSARS-CoV-2感染歴がない被験者におけるVE
- VE2:治験薬接種前から2回目接種後7日以前のSARS-CoV-2感染歴を問わない被験者におけるVE
COVID-19確定例は、COVID-19が疑われる症状(発熱、新たな咳嗽又は咳嗽の悪化、新たな息切れ又は息切れの悪化、悪寒、新たな筋肉痛又は筋肉痛の悪化、新たな味覚又は嗅覚の消失、咽喉痛、下痢、嘔吐)が1つ以上認められ、鼻腔スワブでの核酸増幅検査によりSARS-CoV-2陽性が確認された被験者と定義された。
本試験では、VE1の評価及び無益性による試験の早期中止を目的とした中間解析が当初4回(COVID-19確定例を少なくとも32例、62例、92例及び120例集積後)計画されていたが、運用上の理由により1回目(32例集積時点)は実施せず、試験実施中に3回(COVID-19確定例を少なくとも62例、92例及び120例集積後)に計画変更された。真のVEが30%の場合の試験全体の成功確率を0.025未満とするために、中間解析及び最終解析の有効性の評価の基準値(99.5%及び98.6%)が事前に規定された。
1回目の中間解析は94例集積時点で実施され、当該解析において、真のVE1が30%(FDAガイダンス)において、統計的な成功基準としてVEの第一種の過誤を適切に調整したCIの下限が30%を上回ることが示されていることに基づき設定)を上回る事後確率(>99.99%)は、事前に規定された有効性の基準(99.5%)を上回った。その後、最終解析として計画された少なくとも164例まで、COVID-19確定例が急速に集積されたことから、2回目以降の中間解析は実施されなかった。なお、最終解析終了後も盲検性は維持されている。
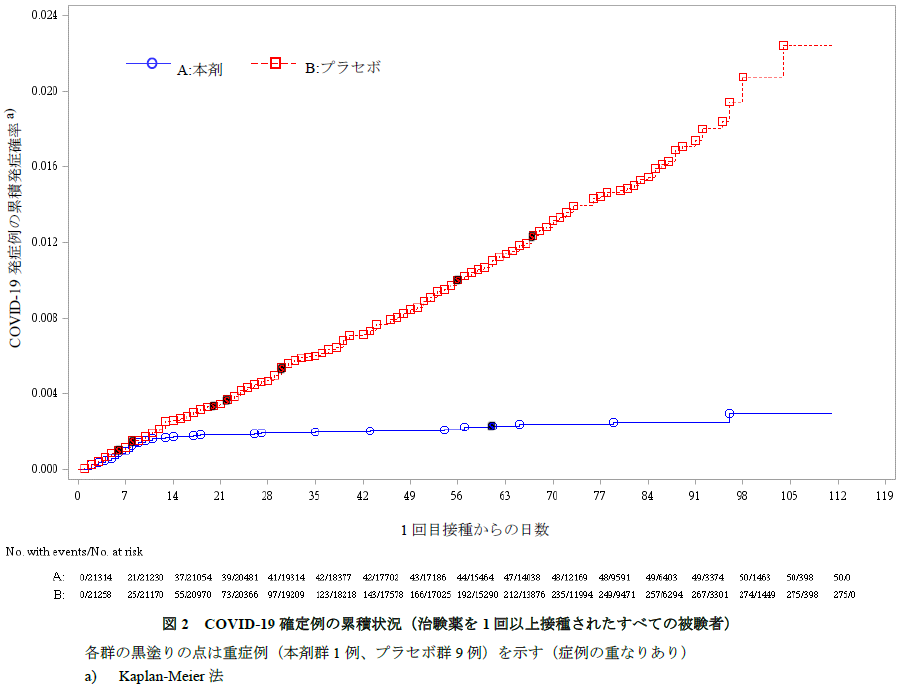
COVID-19確定例が164例以上に到達した時点(最終解析)におけるVE1及びVE2は下表のとおりであった。真のVE1及びVE2が30%を上回る事後確率はいずれも>99.99%であり、事前に規定された有効性の基準(98.6%)を上回った。なお、VE1及びVE2の両側95%CI(Clopper-Pearson法)はそれぞれ[90.0, 97.9]及び[89.6, 97.6]であった。
安全性について、有害事象の重症度は予防ワクチンの臨床試験における毒性評価尺度に関するFDAガイダンスに基づき評価された。安全性解析対象集団におけるデータカットオフ日(2020年11月4日)までの観察期間は、2回目接種から、2週未満14.9%(6,483/43,448例)、2~4週未満5.6%(2,433/43,448例)、4~6 週未満14.9%(6,474/43,448例)、6~8週未満20.7%(8,991/43,448例)、8~10週未満29.1%(12,625/43,448例)、10~12週未満13.0%(5,662/43,448例)、12~14週未満1.8%(780/43,448例)であった。
各観察期間は以下のとおりとされた。
<反応原性解析対象集団のみ>
- 反応原性事象(局所反応(注射部位疼痛、発赤及び腫脹)及び全身反応(発熱、疲労、頭痛、悪寒、嘔吐、下痢、筋肉痛及び関節痛)):治験薬各回接種後7日間(被験者日誌により収集)
<安全性解析対象集団>
- 有害事象(反応原性解析対象集団で治験薬各回接種7日目までに被験者日誌で収集される反応原性事象除く):治験薬1回目接種時から最終接種後1カ月
- 重篤な有害事象:治験薬1回目接種時から最終接種後6カ月
治験薬各回接種後7日間に認められた反応原性事象は下表のとおりであった。
有害事象及び副反応の発現割合は、本剤群26.7%(5,770/21,621例)及び20.7%(4,484/21,621例)、プラセボ群12.2%(2,638/21,631例)及び5.1%(1,095/21,631例)であった。いずれかの群で1%以上に発現した有害事象及び副反応は下表のとおりであった。
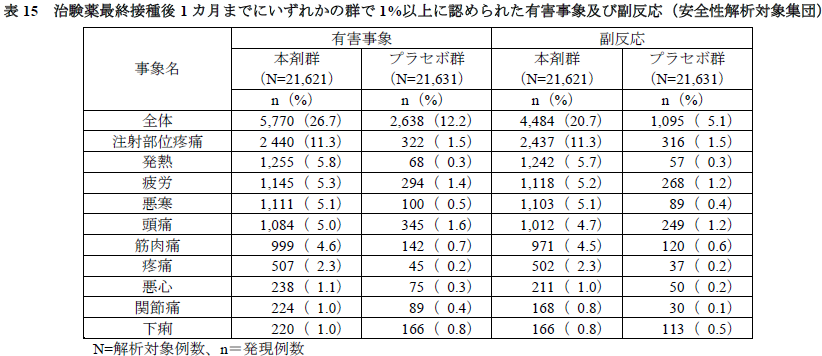
データカットオフ日(2020年11月14日)までに、死亡は本剤群2例(動脈硬化症及び心停止各1例)、プラセボ群4例(原因不明2例、出血性卒中及び心筋梗塞各1例)に認められ、いずれも治験薬との因果関係は否定された。
重篤な有害事象は、本剤群126/21,621例(0.6%)、プラセボ群111/21,631例(0.5%)に認められ、そのうち治験薬との因果関係が否定されなかった有害事象は、本剤群4例(リンパ節症、ワクチン投与関連肩損傷、心室性不整脈、背部痛及び神経根の錯感覚を伴う両下肢の疼痛(MedDRAでコード化されていない事象)各1例)であり、転帰はリンパ節症は未回復、心室性不整脈は回復であり、それ以外の事象は軽快であった。
治験中止に至った有害事象は、本剤群37/21,621例(0.2%)、プラセボ群30/21,631例(0.1%)に認められ、そのうち治験薬との因果関係が否定されなかった有害事象は、本剤群16例、プラセボ群9例に認められた。盲検下で試験継続中であり、接種群ごとの内訳は示されていないが、計25例の内訳(複数の事象が認められた被験者あり)は、下痢及び頭痛各3例、疲労、注射部位疼痛、蕁麻疹及び浮動性めまい各2例、注射部位皮膚炎、回転性めまい、注射部位腫脹、ワクチンアレルギー、眼痛、腹部不快感、筋力低下、四肢痛、リンパ節症、心拍数不整、筋肉痛、口の錯感覚、悪心、頻脈、悪寒、発熱、腹痛、寝汗、片耳難聴、妊娠時曝露及びうつ病各1例、コード化されていない事象2例(ワクチンによる上半身発赤、疲労)であった。転帰はリンパ節症及びうつ病各1例は未回復、妊娠時曝露1 例は転帰不明であり、その他は回復又は軽快であった。:2022年10月24日時点でCDCが重症型の副反応として注意を喚起している「アナフィラキシー」「血栓を伴う血小板減少症候群」「心筋症と心膜炎」「ギラン・バレー症候群」は、この治験(C4591001試験)では認められませんでした。
なお、HIV陽性被験者は主要な安全性解析には含まれず、HIV 陽性被験者の安全性評価は探索的な解析とされた。HIV陽性被験者での有害事象及び副反応の発現割合は本剤群13.1%(13/99例)及び10.1%(10/99例)、プラセボ群10.3%(10/97例)及び0例であった。
機構における審査の概略
「臨床データパッケージ及び審査の方針について」
COVID-19の世界的な流行下において、迅速なSARS-CoV-2ワクチンの開発が求められており、その加速化のためにICMRA、WHO、各国の規制当局は開発についてのガイダンス等を公表している。
PMDAは、令和2年9月2日に「新型コロナウイルス(SARS-CoV-2)ワクチンの評価に関する考え方」を公表し、臨床試験に関しては主に以下の考え方を提示した。
- 感染症予防ワクチンの有効性は、原則として発症予防効果を主要評価項目として評価を行うものであり、COVID-19の発症予防効果について代替となる評価指標が明らかになっていない現状においては、原則として、SARS-CoV-2ワクチン候補の有効性を評価するために、COVID-19の発症予防効果を評価する臨床試験を実施する必要がある。
- SARS-CoV-2については、COVID-19の流行の程度が国・地域によって異なること、ウイルス株が地理的・時間的条件によって異なっていく可能性があること、また、COVID-19が重症化する患者の割合が国・地域によって大きく異なり、その背景については様々な検討がなされていることを踏まえると、SARS-CoV-2ワクチンのベネフィット・リスクの判断については、各国・地域の状況によって異なる可能性がある。その他、民族的要因の差がSARS-CoV-2ワクチンの有効性及び安全性に影響することも考えられる。そのため、海外で発症予防効果を評価するための大規模な検証的臨床試験が実施される場合においても、国内で臨床試験を実施し、日本人被験者において、ワクチンの有効性及び安全性を検討することは、必要性が高いと考える。
- 海外で発症予防効果を主要評価項目とした大規模な検証的臨床試験が実施される場合には、国内で日本人における発症予防効果を評価することを目的とした検証的臨床試験を実施することなく、日本人における免疫原性及び安全性を確認することを目的とした国内臨床試験を実施することで十分な場合がある。
申請者は、本剤の国内臨床試験の計画時点で、海外で発症予防効果を主要評価項目とした大規模な検証的試験を実施中であったこと、本邦のCOVID-19の流行状況を踏まえると発症予防効果を評価することを目的とした国内臨床試験の実施は実施可能性の観点から困難であったことから、国内では免疫原性及び安全性を確認する国内臨床試験を計画・実施した。
海外第Ⅰ/Ⅱ/Ⅲ相試験(海外C4591001試験)及び国内第Ⅰ/Ⅱ相試験(国内C4591005試験)を評価資料として、本申請における臨床データパッケージを構築した。
PMDAは、現時点でCOVID-19の発症予防効果の代替となる評価指標が明らかになっておらず、発症予防効果と免疫原性との関連は明確ではないものの、迅速なSARS-CoV-2 ワクチンの開発が求められている状況等を考慮すると、本剤の有効性については、海外の検証的試験(海外C4591001試験)の成績に基づき評価し、それに加えて国内臨床試験成績から日本人の免疫原性及び安全性を確認することで、日本人における本剤の有効性及び安全性を評価することとした。:パンデミックの最中であったことから(緊急対応として)、日本人における有効性は、発症予防効果のサロゲートマーカーとして適切かどうか不明であった免疫原性を基づいて評価されました。
「有効性について」
PMDAは、提出された試験成績及び以下の検討を踏まえ、海外C4591001試験成績から本剤のCOVID-19発症予防効果は示され、海外C4591001試験及び国内C4591005試験で得られた免疫原性データの結果から、日本人においても同様の有効性が期待できると判断した。
ただし、本剤の長期の有効性及びSARS-CoV-2変異株に対する有効性については、現時点で得られている情報からは不明であることから、製造販売後に引き続き情報収集し、新たな知見が得られた場合には医療現場に情報提供する等、適切に対応する必要があると考える。
:上記のとおり、この審査報告書を作成した時点で、本剤の長期の有効性については不明、と述べられていますので、このブログでは有効性については省略しようと思いますが、VE(ワクチン効果)についてだけコメントします。
海外C4591001試験の第Ⅱ/Ⅲ相パートのデータをまとめた下の図表が示すとおり、本剤の1回接種以降全期間で82%、2回目接種後7日以降で94.8%のVEが記録されています。「日本人における有効性について」の項で、申請者は「国内C4591005試験の日本人被験者で、海外C4591001試験と同様のSARS-CoV-2血清中和抗体価の上昇が認められていること、複数の国、人種、民族が組み入れられた海外C4591001試験において有効性が示されたことを踏まえると、日本人においても海外C4591001試験と同様の本剤の有効性は期待できると考える」と述べています。
しかしながら、2022年10月17日時点で人口の約8割が2回以上のワクチン(他社製品も含まれますが)を接種されているにもかかわらず、5回目の接種が開始されている状況であることを考えると、本剤を含めmRNAワクチンの有効性に人種差があるのか、あるいは(人種差はないのであれば)効果に持続性はあまりないのかなぁと思ってしまいます(個人的見解です)。
「安全性について」
PMDAは、本剤の安全性について、以下のように判断した。
- 提出された資料における本剤の安全性情報は、海外C4591001試験の第Ⅱ/Ⅲ相パートでは2回目接種後1~3 カ月を中心とするデータ(観察期間が2回目接種後4週以上12週未満の被験者の割合は77.7%(33,752/43,448 例))及び国内C4591005試験では2回目接種後1カ月のデータであり、現時点で本剤接種後長期の十分な安全性データは得られていないことには留意が必要である。その上で、提出された資料に基づき以下の検討を行った結果、現時点で本剤の承認の可否に影響する重大な懸念は認められていない。
- 以下の情報については製造販売後に情報収集を行い、本剤の情報については、継続中の臨床試験や海外の情報を含め、速やかに情報収集し、得られた知見に応じて追加の注意喚起や情報提供の要否を検討する等、適切に対応する必要があると考える。
- 本剤接種後の長期の安全性
- COVID-19 の重症化リスクとなる基礎疾患及び背景を有する被接種者の安全性
- 本剤の疾患増強リスク
安全性プロファイルについて
申請者の説明は、
①有害事象について
- 海外C4591001試験の第Ⅱ/Ⅲ相パートでは、最初に組み入れられた8,214例(本剤群4,108例、プラセボ群4,106例)について、事前に規定した局所反応(注射部位疼痛、発赤及び腫脹)及び全身反応(発熱、疲労、頭痛、悪寒、嘔吐、下痢、筋肉痛及び関節痛)を、治験薬各回接種後7日間、被験者日誌により重点的に収集し、これらを反応原性事象として解析した。
- 結果は下表のとおりであり、本剤群の多くの被験者で1回目又は2回目接種後7日間のいずれかに、局所反応及び全身反応が認められ(それぞれ84.7%及び77.4%)、発現割合はいずれもプラセボ群よりも高かった。各事象の発現割合は、嘔吐及び下痢は本剤群とプラセボ群で同程度であったが、それ以外は本剤群でプラセボ群よりも高く、また、本剤群ではほとんどの事象が10%以上に認められた。Grade 3以上が1%以上に認められた事象は、疲労、頭痛、筋肉痛、悪寒、注射部位疼痛であった。
- 発熱(38℃以上)はGrade分類されていないが、本剤群の体温別の発現割合は38.0~38.4℃は9.2%(378例)、38.5~38.9℃は4.1%(167例)、39.0~40.0℃は0.9%(35例)、40.0℃超は0.0%(2例)であった。
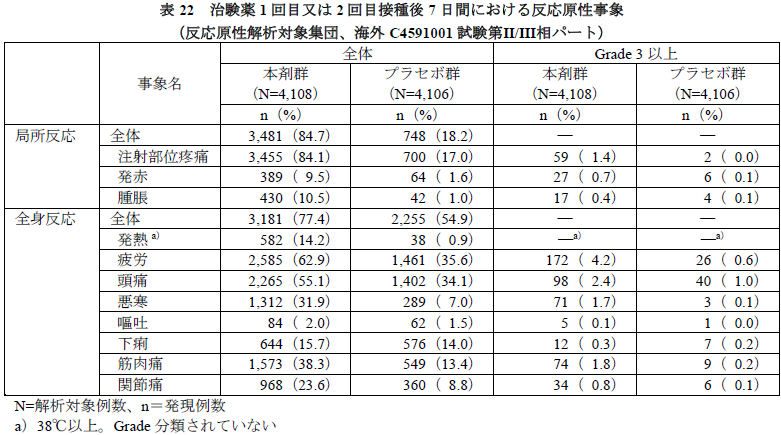
- 局所反応は多くが治験薬接種当日~3日目に認められ、一部約1カ月間持続した症例や転帰不明の症例もあるが、ほとんどが1~2日後に消失した。全身反応は多くが治験薬接種2~3日目に認められ、一部約1カ月間持続した症例や転帰不明の症例もあるが、ほとんどが1日後に消失した。
- 治験薬接種に伴う症状の治療に対する解熱鎮痛剤の使用は許容されており(予防投与は許容されない)、解熱鎮痛剤を1回以上使用した被験者は本剤群46.5%(1,909例)、プラセボ群19.7%(810例)であった。
- 治験薬最終接種後1カ月までの有害事象(反応原性解析対象集団における治験薬各回接種後7日間の被験者日誌で収集された反応原性事象除く)の発現割合は、本剤群26.7%(5,770/21,621例)、プラセボ群12.2%(2,638/21,631例)であった。
- 本剤群の1%以上に認められた有害事象は、注射部位疼痛、発熱、疲労、悪寒、頭痛、筋肉痛、疼痛、悪心、関節痛及び下痢であり、疼痛以外は反応原性事象として規定した事象であり、これらの事象の多くが接種後7日間に認められ、本剤との因果関係ありと判断された。
- MedDRA器官別大分類「神経系障害」に分類される有害事象は本剤群5.9%(1,277/21,621 例)、プラセボ群2.3%(501/21,631例)(以下、同順)に認められ、事象別の発現割合は、頭痛(5.0%、1.6%)、浮動性めまい(0.3%、0.2%)、錯感覚(0.1%、0.1%)、片頭痛(0.1%、0.0%)、嗜眠(0.1%、0.0%)等であった。
- ギラン・バレー症候群及び急性散在性脳脊髄炎は認められなかった。:この治験では、ギラン・バレー症候群は観察されませんでした。
- リンパ節症は、本剤群0.3%(70/21,621例)、プラセボ群0.0%(7/21,631例)に認められ、このうち本剤群50例、プラセボ群4例で治験薬との因果関係ありと判断された。
- リンパ節症は大部分の症例で腕又は頚部に発現した。多くは治験薬接種後2~4日以内に発現したが、本剤群12例及びプラセボ群3例で接種後8日目以降(最長98日目)に認められた。
- また、本剤群1例で接種後30分以内に認められた。本剤群1例は重篤例であり、因果関係はありとされ、転帰は未回復であった。発現状況からリンパ節症については、本剤により発現した反応原性事象と判断し、添付文書で注意喚起を行う。
- 国内C4591005試験では治験薬各回接種後7日間の反応原性事象は安全性解析対象集団全例で評価され、本剤群の多くの被験者で1回目又は2回目接種後7日間のいずれかに、局所反応及び全身反応(それぞれ91.6%及び78.2%)が認められ、発現割合はプラセボ群よりも高かった(下表)。
- 各事象の発現割合は、嘔吐は本剤群とプラセボ群で同程度であったが、それ以外は本剤群でプラセボ群よりも高く、また、本剤群ではほとんどの事象が10%以上に認められた。Grade 3以上の事象は、疲労、注射部位疼痛、頭痛、悪寒及び関節痛に認められた。
- 発熱(37.5℃以上)はGrade分類されていないが、本剤群の体温別の発現割合は37.5~37.9℃は17.6%(21例)、38.0~38.4℃は9.2%(11例)、38.5~38.9℃は8.4%(10例)、39.0~40.0℃は0.8%(1例)、40.0℃超は0例であった。
- 国内C4591005試験の発熱の発現割合(36.1%)は、海外C4591001試験の第Ⅱ/Ⅲ相パート(14.2%)よりも高かったが、国内C4591005試験では、発熱の定義を37.5℃以上とし、海外C4591001試験で収集した発熱(38℃以上)よりも広い体温の範囲を収集し、37.5℃~37.9℃の発熱が多く収集されたことが、国内C4591005試験で発現割合が高くなった要因と考えた。
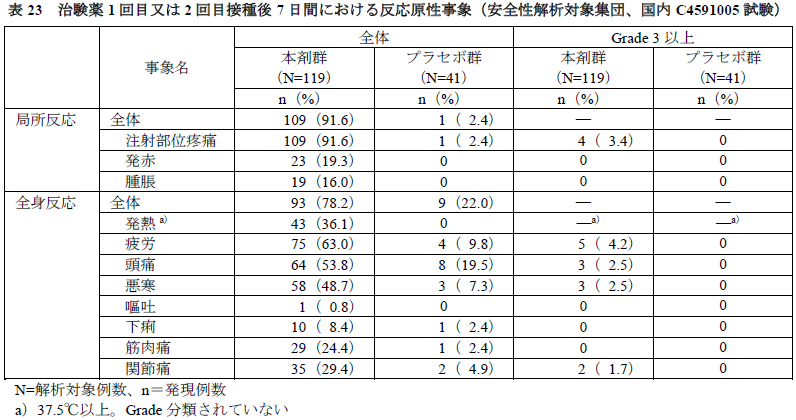
- 局所反応は多くが治験薬接種当日~3日目に認められ、ほとんどが発現から1~3.5日後に消失した。全身反応は多くが接種2~4日目に認められ、ほとんどが発現から1日後に消失した。なお、解熱鎮痛剤を1回以上使用した被験者は本剤群37.8%(45例)、プラセボ群4.9%(2例)であった。
- 治験薬最終接種後1カ月までの有害事象の発現割合(治験薬各回接種後7日間の被験者日誌で収集された反応原性事象を除く)は、本剤群10.1%(12/119例)、プラセボ群7.3%(3/41例)であり、本剤群で2例以上に認められた有害事象は上咽頭炎(3例)及び頭痛(2例)であった。なお、神経系障害に分類される有害事象は頭痛(2例)のみであり、リンパ節症は認められなかった。
②接種回別及び年齢別の有害事象について
- 反応原性事象について、接種回別及び年齢別の結果を下表(海外C4591001試験 第Ⅱ/Ⅲ相パート)及び(国内C4591005試験)に示す。
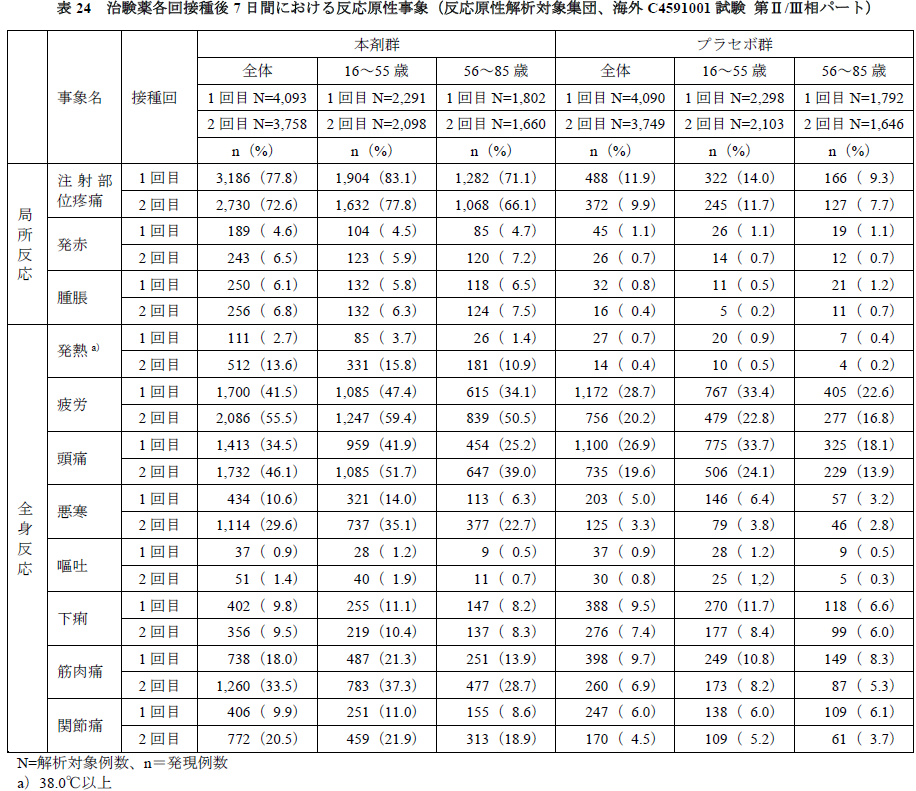
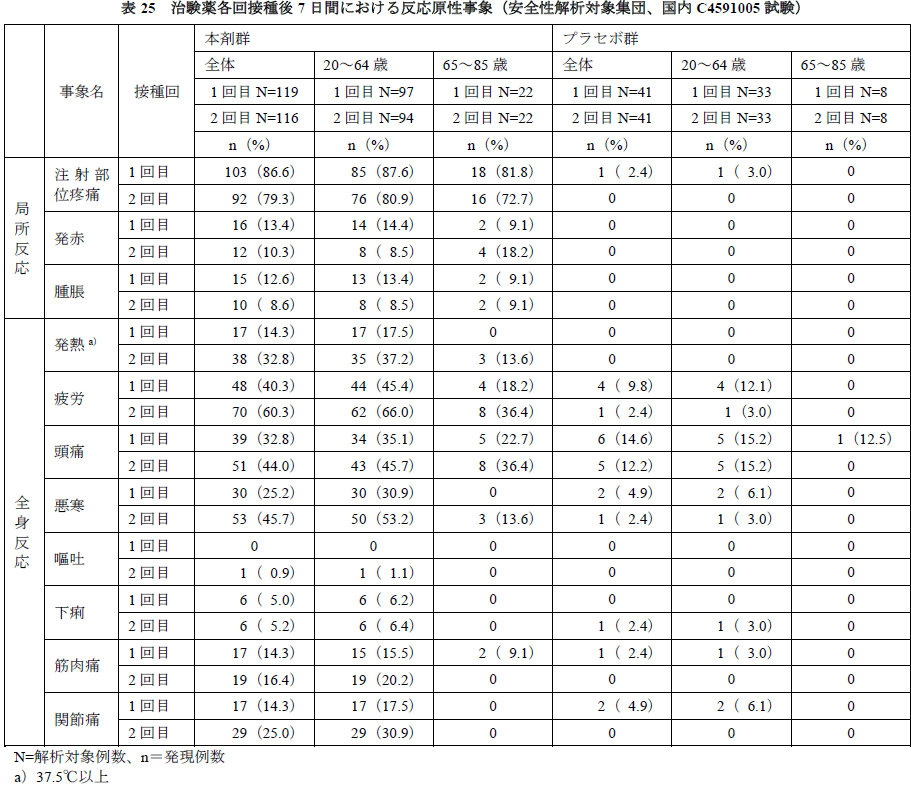
- 海外C4591001試験第Ⅱ/Ⅲ相パートについて、接種回別では、局所反応の各事象や、全身反応のうち嘔吐及び下痢の発現割合は各回接種後で同程度であったが、それ以外の全身反応は1回目接種後よりも2回目接種後で発現割合が高かった。
- Grade 3以上の事象の発現割合は、1回目接種後ではいずれも1%未満であったが、2回目接種後では疲労(3.8%)、頭痛(2.0%)、筋肉痛(1.7%)及び悪寒(1.6%)で1%以上に認められた。
- 年齢別では、各事象の発現割合及びGrade 3以上の事象の発現割合はほとんどが非高年齢層で高年齢層よりも発現割合が高く、高年齢層で顕著に発現割合が高い事象は認められなかった。
- 発熱(38.0℃以上)について、本剤群の発現割合は1回目接種後よりも2回目接種後で高く、高年齢層よりも非高年齢層で高かった。そのうち38.9℃超は1回目接種後0.2%(8例:非高年齢層6例、高年齢層2例)、2 回目接種後0.8%(32例:非高年齢層27例、高年齢層5例)、40.0℃超は2例(1回目接種後・高年齢層及び2回目接種後・非高年齢層各1例)に認められた。発現日(中央値)は接種2日目、持続期間(中央値)は1日であり、接種回別及び年齢層別で同様であった。
- また、治験薬最終接種後1カ月までの有害事象(反応原性解析対象集団における治験薬各回接種後7日間の被験者日誌で収集された反応原性事象除く)については、ほとんどが反応原性事象として規定した局所反応又は全身反応であり、接種回別及び年齢別の発現傾向についても反応原性事象の結果と同様の傾向であった。なお、16~17歳の被験者での有害事象発現割合は、本剤群11.6%(16/138例)、プラセボ群4.8%(7/145例)であり、本剤群で認められた事象は、反応原性事象として規定した事象がほとんどであった。
- 国内C4591005試験の接種回別及び年齢別の結果については、海外C4591001試験第Ⅱ/Ⅲ相パートと同様の傾向であった。
③重篤な有害事象について
- 海外C4591001試験の第Ⅰ相パート(データカットオフ日2020年8月24日)に、死亡及び重篤な有害事象は認められなかった。なお、本剤30μg群の2回目接種後の追加の観察期間中に末梢神経損傷(当初は神経炎として報告)1例が報告されたが、治験薬との因果関係は否定された。
- 海外C4591001試験の第Ⅱ/Ⅲ相パート(データカットオフ日2020年11月14日)に、重篤な有害事象は本剤群126/21,621例(0.6%)、プラセボ群111/21,631例(0.5%)に認められ、治験薬との関連が否定されなかった事象は、本剤群4例(リンパ節症、ワクチン投与関連肩損傷、心室性不整脈、背部痛及び神経根の錯感覚を伴う両下肢の疼痛各1例)であり、転帰はリンパ節症は未回復、心室性不整脈は回復であり、それ以外の事象は軽快であった。死亡例は、本剤群2例(動脈硬化症及び心停止各1例)、プラセボ群4例(原因不明2例、出血性卒中及び心筋梗塞各1例)に認められ、いずれも治験薬との因果関係は否定された。
- データカットオフ日以降2020年12月29日までに、死亡例は10例(盲検下で試験継続中であり接種群不明:心肺停止2例、心停止、うっ血性心不全、高血圧性心疾患、動脈硬化症、肺炎、COVID-19、COVID-19肺炎、胆嚢機能低下、敗血症性ショック、大動脈破裂、糖尿病各1 例(重複含む))に認められ、重篤な有害事象は91例に認められたが、すべて治験薬との因果関係は否定された。国内C4591005試験では死亡及び重篤な有害事象は認められなかった(データカットオフ日2021年1月5日)。
以上より申請者は、国内外の臨床試験の結果において、
- 被験者の多くに反応原性事象が認められ、非高年齢層で高年齢層よりも多く認められる傾向であったが、ほとんどの事象は軽度又は中等度であった。
- 接種後短期間で消失していた。
- 死亡及び重篤な有害事象の発現割合は低く、ほとんどで本剤群との因果関係は否定されている。
- 16歳以上の被接種者において本剤の安全性プロファイルに重大な懸念は認められない。
したがって、忍容性は確認されたと考える。
この説明に対してPMDAは、
提出された国内外の臨床試験成績において、
- 本剤接種後長期の十分な安全性データは得られていないことには留意が必要である。
- その上で、現時点の情報において、被験者の多くに反応原性事象として収集された局所反応及び全身反応が認められていたものの、ほとんどが軽度又は中等度であり、回復性が認められている。
- 国内外の安全性プロファイルに大きな差異は認められていない。
- その他の有害事象の発現状況や年齢別の発現状況。
以上から判断すると、本剤の承認の可否に影響する重大な懸念は認められていないと考える。
ただし、多くの被験者に認められた全身反応は日常生活に影響を及ぼす可能性があり、Grade 3の全身反応及び37.5℃以上の発熱も一定の割合で認められていること、並びに1回目接種後よりも2回目接種後で発現割合が高い事象が認められていることは、本剤の被接種者にとって重要な情報である。
これらの情報については発現時期や持続期間等も含めて、医療従事者や被接種者等に対して適切に情報提供する必要がある。また、本剤接種後長期の安全性情報については、製造販売後に引き続き情報収集する必要がある、と回答した。
ショック、アナフィラキシーについて
本剤の海外での使用許可後又は製造販売後に、重篤な過敏症反応が報告されていることから、PMDAは、本剤接種後の過敏症反応の発現状況について申請者に説明を求め、申請者は以下のように説明した。
- 海外C4591001 試験の第Ⅱ/Ⅲ相パートでは、MedDRA器官別大分類「免疫系障害」に分類される事象は本剤群0.1%(26/21,621例)、プラセボ群0.1%(22/21,631例)に認められ、このうち本剤群6例(免疫反応5例、薬物過敏症1例)、プラセボ群1例(ワクチンアレルギー)で治験薬との因果関係が否定されなかった。
- 本剤群で認められた免疫系障害に分類される事象はいずれも軽度又は中等度であり、1回目又は2回目の接種当日又は翌日に発現した。また、MedDRA SMQ(狭義)で血管浮腫及び過敏症に該当する事象を抽出したところ、本剤群でそれぞれ0.1%(25/21,621例)及び0.7%(144/21,621例)、プラセボ群でそれぞれ0.1%(23/21,631例)及び0.6%(120/21,631例)であった。本剤接種直後(30分以内)の即時型アレルギー反応は認められなかった。
- 重篤例は、本剤群2例(アナフィラキシー反応及び薬物過敏症各1例)、プラセボ群1例(アナフィラキシーショック)に認められ、いずれも治験薬との因果関係は否定されている。
- 海外C4591001試験の第Ⅱ/Ⅲ相パートでは、非重篤なアレルギーの既往がある被験者が本剤群5,839例、プラセボ群5,834例(うち、アナフィラキシーの既往がある被験者は本剤群15例、プラセボ群22例)組み入れられた。これらの被験者で治験薬との関連のあるアレルギー関連の事象は、本剤群1例(薬物過敏症及び蕁麻疹)、プラセボ群1例(ワクチンアレルギー及び咽頭腫脹)に認められ、いずれも中等度であり、転帰は回復であった。国内C4591005試験ではアレルギー関連の事象は認められなかった(データカットオフ日2021年1月5日)。
- 臨床試験及び海外における使用許可後又は製造販売後の安全性情報でショックを含むアナフィラキシーが認められていることから、ショック、アナフィラキシーについては、添付文書で注意喚起を行う。
この説明に対してPMDAは、
臨床試験及び海外における使用許可後又は製造販売後の安全性情報におけるショック、アナフィラキシー等の発現状況を確認し、これらについて添付文書で注意喚起を行うとの申請者の説明を了承した。
また、接種前には、被接種者の既往歴等を確認し、接種後一定時間は被接種者の観察を行うことが望ましく、異常が認められた場合には適切な処置を行う必要がある旨についても、情報提供する必要がある、と回答した。
顔面麻痺(ベル麻痺)について
海外C4591001試験の第Ⅱ/Ⅲ相パート
で顔面麻痺が本剤群4例に認められていることについて、申請者申請者の説明は、
- 海外C4591001試験の第Ⅱ/Ⅲ相パートの本剤群4例で認められた顔面麻痺について、2例が治験薬との関連ありと判断され、いずれも軽度又は中等度であり、転帰は回復又は消失であった。
- 国内C4591005試験において顔面麻痺は認められなかった(データカットオフ日2021年1月5日)。また、海外での使用許可後又は製造販売後の自発報告において顔面麻痺は21例報告された。
- 顔面麻痺の発症率は、申請者が保有する米国の電子健康記録データベースでは、10万人年当たり77人であり、本剤の臨床試験での発現割合は若干高いものの、予測の範囲内であった。
- 一方、複数の文献報告によれば発症率は10万人年当たり15~30人であり、本剤の臨床試験での発現例数は文献報告の発症率に基づき予想される例数よりも4.3倍高かった。
現時点では本剤と顔面麻痺との因果関係は不明であり、引き続き検討を行うが、顔面麻痺については添付文書で注意喚起を行う。
PMDAは、臨床試験及び海外における使用許可後又は製造販売後の安全性情報における顔面麻痺の発現状況を確認し、これについて添付文書で注意喚起を行うとの申請者の説明を了承した。
基礎疾患を有する人における安全性について
PMDAは、SARS-CoV-2ワクチン接種の必要性が高いと考えられるCOVID-19の重症化リスクが高い基礎疾患を有する被接種者での本剤の安全性について、申請者に説明を求めた。
申請者の説明は、
- 海外C4591001試験の第Ⅱ/Ⅲ相パートの結果から、治験参加時に基礎疾患(Charlson Comorbidity Indexに示される疾患)を有する被験者及びCOVID-19の重症化リスクとされる肥満(BMI 30kg/m2以上)の被験者について、事後解析を実施した。
- 解析に含まれる基礎疾患を有する被験者(8,978例)には、慢性肺疾患3,443例、糖尿病:慢性合併症なし3,368例、慢性合併症あり237例、悪性疾患1,561例等が含まれ、新型コロナウイルス感染症COVID-19診療の手引きでCOVID-19の重症化リスクが高いとされる基礎疾患が含まれていた。また、AIDS/HIVも197例含まれていた。これらの集団での反応原性事象は下表のとおりであった。
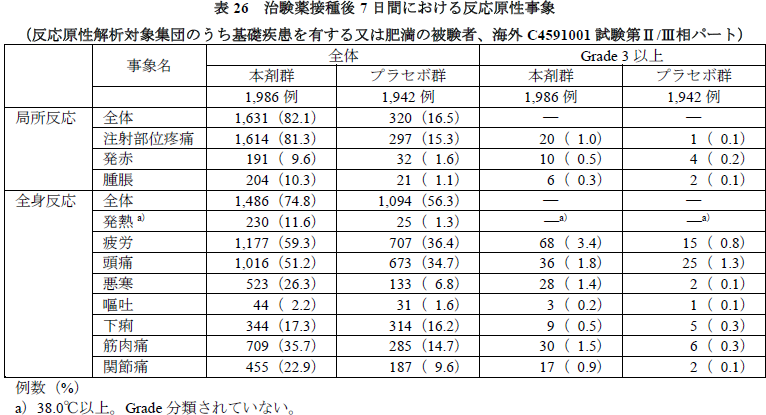
- 有害事象の発現割合は本剤群25.0%(2,172/8,697例)、プラセボ群13.0%(1,125/8,641例)であり、治験薬との因果関係ありとされた事象は、本剤群18.1%(1,575/8,697例)、プラセボ群5.1%(439/8,641例)であった。
- 本剤群の1%以上に認められた有害事象は、注射部位疼痛、発熱、疲労、悪寒、頭痛、筋肉痛、疼痛、悪心、関節痛、下痢であった。以上の解析結果は全体集団の結果と同様であった。現在、厚生労働省で新型コロナウイルスワクチンの接種順位の上位に位置付ける基礎疾患について検討されている。
- 現時点で新型コロナウイルス感染症COVID-19診療の手引きにおいては、COVID-19の重症化リスクが高い基礎疾患及び背景として、悪性腫瘍、慢性閉塞性肺疾患、慢性腎臓病、2型糖尿病、高血圧、脂質異常症、BMI 30kg/m2以上の肥満、固形臓器移植後の免疫不全等が挙げられている。
- 海外C4591001 試験の第Ⅱ/Ⅲ相パートの結果における基礎疾患等を有する被験者の事後解析にはこれらの基礎疾患を有する情報も一部含まれるものの、現時点で十分な情報は得られていない。
- そのため製造販売後にはCOVID-19 の重症化リスクが高い基礎疾患を有する被接種者における本剤の安全性について情報収集する予定である。
この説明に対してPMDAは、
- 海外C4591001 試験の第Ⅱ/Ⅲ相パートの結果から、基礎疾患を有する又は肥満の被験者における安全性は全体集団と同様であったことを確認した。
- ただし、臨床試験で組み入れられた基礎疾患は比較的安定した状態であり、製造販売後には様々な状態の基礎疾患を有する人に接種されると想定される。:医薬品は一旦上市されると、開発段階では想定できないような状況で使用されることがあるので、どの医薬品も市販後調査が必要となります。
以上より、使用実態下における情報収集は重要と考えることから、製造販売後には、本剤の必要性が高いと考えられるCOVID-19 の重症化リスクが高い基礎疾患を有する被接種者における本剤の安全性について、情報収集するとの申請者の説明を了承した。
妊婦に対する安全性について
申請者の説明は、
- 本剤の臨床試験では、妊婦は除外基準に規定されていたが、海外C4591001試験の第Ⅱ/Ⅲ相パートで、23例に妊娠が報告され、そのうち9例は妊娠を理由に治験中止された。これらの被験者の妊娠の転帰については現時点で情報は得られておらず、引き続き追跡する。
- 海外での使用許可後又は製造販売後の自発報告(報告対象期間2020年12月1日~同年12月31日)において妊婦への投与は28例に確認され、特段の懸念は認められていない。
- 生殖発生毒性試験では特段の懸念は認められていない。
以上より、妊婦に対しては予防接種上の有益性が危険性を上回ると判断される場合に接種することは可能と考える。
PMDAは、申請者の説明を了承し、臨床試験で本剤接種された妊婦の妊娠の転帰や製造販売後の情報から、新たな知見が得られた場合には、追加の注意喚起の要否を検討する等、適切に対応する必要があると考える、と回答した。
疾患増強リスクについて
申請者の説明は、
- 薬理での検討では、動物及びヒトにおける本剤接種後のサイトカイン産生の検討等から、本剤接種による疾患増強リスクは低いと考えられた。:Th2型免疫反応が疾患増強につながることを前提とした見解ですね。
- 臨床試験では、COVID-19に罹患した患者が少ないこと、また、疾患増強リスクの評価については長期の観察が必要と考えられるが、現時点で得られている情報は治験薬2回目接種後1~3カ月までのデータが中心(観察期間が治験薬2回目接種後4週以上12週未満の被験者の割合は77.7%(33,752/43,448例))であることから、本剤による疾患増強リスクを評価することは困難である。
- 現時点で本剤のヒトでの疾患増強リスクは不明である。
以上より、製造販売後に引き続き情報収集する。
PMDAは、申請者の説明を了承し、本剤のヒトでの疾患増強リスクについては、製造販売後に引き続き海外の情報を含めて情報収集し、新たな知見が得られた場合には速やかに情報提供を行うことが適切と考える、と回答した。
海外における使用許可後又は製造販売後の安全性情報について
申請者の説明は、
- 本剤の第1回Summary Monthly Safety Report(2021年1月13日付け)における報告対象期間(2020年12月1日~同年12月31日)に、3,615例の自発報告がされ、12の有効な安全性シグナル(アナフィラキシー、注射部位発赤、注射部位腫脹、倦怠感、悪心、嘔吐、下痢、過敏症、不眠症、注射部位そう痒感、四肢痛、顔面麻痺)が検出された。
- アナフィラキシーは重要な特定されたリスク、注射部位発赤、注射部位腫脹、倦怠感及び悪心は特定されたリスク(重要なリスクではない)、嘔吐及び下痢はリスクではないと判断され、残りは継続して評価することとされた。
評価の結果、当該報告対象期間における本剤のベネフィット・リスクプロファイルは良好と判断している。
この説明に対してPMDAは、
本剤は2020年12月から接種開始され、死亡例を含む重篤な事象が報告されているものの、現時点では因果関係が明らかではないこと、海外での安全性情報は今後蓄積される状況であることから、得られた情報に基づき、本剤の安全性について継続的に評価し、追加の注意喚起や情報提供の要否を検討する等、適切に対応する必要があると考える、と回答した。
「臨床的位置づけについて」
本剤の臨床的位置付けについて、PMDAの考えは以下のとおり。
- 2021年1月20日時点の、本邦でのSARS-CoV-2の感染者数(PCR検査陽性者数)の累計は332,231例、うち重症例は71,129例、死亡は4,547例である。
- 無症状感染者はすべてを把握できないため、無症状者を含めた感染者数はさらに多いと推測される。年代別の感染数は20代が最も多く、次いで30代、40代、50代の順に多いが、死亡者数や重症者数は60代以上に多い。
- SARS-CoV-2曝露から発症までの潜伏期間は1~14日間で、通常は5日程度で発症することが多い。また、症状が発現する前から感染性があり、発症から間もない時期の感染性が高いことが市中感染の原因とされている。
- 本邦では、2020年5月7日にSARS-CoV-2による感染症の効能・効果で抗ウイルス薬であるレムデシビルが治療薬として承認された。また、デキサメタゾンは既承認の効能・効果の範囲で使用可能であり、その他、医療現場では重症度や症状に応じて各種治療薬が用いられているが、これらの治療を行っても感染者数、重症例及び死亡例は増加している。
- また、COVID-19との因果関係は明らかとなっていないが、一部の感染者ではウイルス消失後も、嗅覚障害、味覚障害、呼吸困難、脱毛等の症状が遷延するという報告もある。
- 2021年1月時点で、本邦で感染者数の増加が続いており、医療体制がひっ迫している状況であること、COVID-19を発症すると、重症化や死亡の転帰となる場合もあることを踏まえると、COVID-19の発症予防は極めて重要である。「新型コロナウイルス感染症対策分科会での議論と政府としての中間とりまとめ」によると、「新型コロナウイルス感染症による死亡者や重症者の発生をできる限り減らし、結果として新型コロナウイルス感染症のまん延の防止を図る」ことがワクチン接種の目的とされているが、本邦でCOVID-19の予防を目的として承認されているワクチンはない。
- 海外C4591001試験の第Ⅱ/Ⅲ相パートの結果から、本剤のCOVID-19の発症予防効果は示され、国内C4591005試験で海外C4591001試験と同程度以上の血清中和抗体価の上昇が確認されたことから、日本人に対しても同様のCOVID-19の発症予防効果が期待できると考えられ、安全性及び忍容性についても承認の可否に影響する懸念はないと考えられた。
- 本剤接種後の長期の有効性及び安全性や重症化抑制効果は現時点では不明であり、変異株に対する本剤の有効性に不確実性はあるものの、本剤の接種によりCOVID-19の発症予防効果が期待でき、国内の発症者数の低減につながることが期待できる。
- 2021年1月時点で感染者数が増加し医療体制がひっ迫している。
以上を踏まえると、本邦初のCOVID-19に対する予防ワクチンとして、本剤を接種可能とすることは意義があると考える。
「効能・効果について」
PMDAは、本剤の効能・効果について、
- 海外C4591001試験の第Ⅱ/Ⅲ相パートの結果から、本剤のCOVID-19の発症予防効果は示され、国内C4591005試験で海外C4591001試験と同程度以上の血清中和抗体価の上昇が確認されたことから、日本人に対しても同様のCOVID-19の発症予防効果は期待できると判断した。
- 「新型コロナウイルス(SARS-CoV-2)ワクチンの評価に関する考え方」の記載、既承認のワクチンの効能・効果等を踏まえると、本剤の効能・効果は、申請時の効能・効果のとおり、「SARS-CoV-2による感染症の予防」とすることが適切と判断した。
「用法・用量について」
申請者の説明は、
- 用量設定試験として実施した海外C4591001試験の第Ⅰ相パートでは、本剤10、20又は30μgを21日間隔で2回接種したときの安全性、忍容性及び免疫原性について検討し、結果は以下のとおりであった。
- 本剤接種後のSARS-CoV-2血清中和抗体価について、いずれの用量でも1回目接種後21日目の上昇はわずかであったが、2回目接種後7日目以降に顕著な上昇が認められた。
- COVID-19の重症化リスクが高いと考えられる高年齢層集団において、中和抗体価は20μg群よりも30μg群で高値であった。
- 安全性については、いずれの用量でも懸念は認められなかった。
- 以上の結果から、第Ⅱ/Ⅲ相パートで検討する本剤の用法・用量は1回30μgを21日間隔(許容期間は19~23日)で2回、筋肉内接種することと設定し、試験を実施した。その結果、本剤の有効性が確認され、安全性及び忍容性についても許容可能と判断した。
- なお、第Ⅱ/Ⅲ相パートにおける、治験薬1回目接種以降のCOVID-19累積発症確率の推定結果は、1回目接種後14日目までは本剤群とプラセボ群で同様に推移し、その後、プラセボ群は増加する一方で本剤群はほとんど増加せず、2回目接種以降で本剤群とプラセボ群とで大きな差が認められた。1回接種のみの有効性については検討していないが、第Ⅰ相パートにおいて中和抗体の誘導には2回接種が必要であったことも踏まえると、1回接種のみでは効果持続性の観点から十分でないと考える。
- また、1回目接種と2回目接種の間隔について、第Ⅱ/Ⅲ相パートでは21日間隔(許容期間は19~23日)として設定していたが、有効性の解析は1回目接種から19~42日後に2回目接種された被験者も含む集団で解析を行うことと事前に規定しており、当該集団で本剤のCOVID-19の発症予防効果が示された。
- 有効性評価可能集団のうち、1回目接種から24~42日後に2回目接種された被験者は、本剤群18,198例中616例、プラセボ群18,325例中659例が含まれていた。これらの集団における2回目接種後7日目以降のCOVID-19確定例は本剤群1例、プラセボ群4例であり、VE[両側95%CI](治験薬接種前から2回目接種後7日以前にSARS-CoV-2感染歴がない被験者)は73.3[-170, 99.5]%であった。
- 例数が少なく、確定的な評価は困難であるが、接種間隔が24~42日間であった集団でも有効性は期待できると考える。
- 国内C4591005試験でも、海外C4591001試験の第Ⅱ/Ⅲ相パート同じ用法・用量を設定して試験実施し、免疫原性の結果から日本人でもCOVID-19の発症予防効果は期待でき、安全性及び忍容性について、日本人特有の懸念は認められなかった。
以上より申請者は、これらの臨床試験の結果に基づき本剤の用法・用量を設定することは可能と考えた。本剤1バイアルを日局生理食塩液1.8mLで希釈した場合、本剤30μgに相当する容量は0.3mLであることから、申請用法・用量は、「日局生理食塩液1.8mLにて希釈し、通常、1回0.3mLを合計2回、3週間の間隔で筋肉内に接種する」と設定した。
この説明に対してPMDAは、
本剤の有効性及び安全性に関する検討結果から、用法・用量について、本剤30μgに相当する、生理食塩液1.8mLにて希釈後0.3mLを1回分として、合計2回、3週間間隔で筋肉内接種と設定することは可能と判断した。なお、1回接種のみでの有効性及び接種間隔を24日以上に延長した場合の有効性については十分に確立していないことから、臨床試験の設定に基づき3週間間隔で2回接種とすることが適切と考える、と回答した。:一般的に(免疫学的に)、ワクチンによる免疫誘導は2回接種で十分と考えられるため、当初は2回接種を前提に開発を進めたと思われますが、2022年5月末から本剤の4回目の投与が開始されたという現実を勘案すると、本剤の有効性の評価について疑問を感じると同時に、新型コロナウイルスの変異株に対するmRNAワクチンの有効性の評価方法について深い議論が必要と考えます。
「接種対象年齢について」
申請者の説明は、
- 国内C4591005試験では、20~85歳を対象として実施し、日本人での安全性、忍容性及び免疫原性を評価した。
- 一方、海外C4591001試験の第Ⅱ/Ⅲ相パートにおいて、16歳以上の被験者での有効性及び安全性が確認され、年齢層別の解析においても臨床的に懸念となるような結果は認められなかったことから、本邦においても、16歳以上を接種対象とすることは可能と考える。
- なお、海外C4591001試験の第Ⅱ/Ⅲ相パートでは、試験実施中に12~15歳の小児についての免疫原性、安全性及び忍容性を検討する計画を追加している。これらの年齢層の被験者は有効性解析において一部のデータが含まれているものの、現時点で十分な結果は得られていない。したがって、小児を対象とした開発計画については、当該結果を踏まえて検討する。
この説明に対してPMDAは、
- 国内C4591005試験では20~85歳が対象とされ、日本人においては、16~19歳におけるデータは得られていないものの、以上の申請者の説明、国内C4591005試験と海外C4591001試験における20歳以上の免疫原性及び安全性プロファイルに大きな差異は認められなかった。
- 現時点の本邦でのSARS-CoV-2の流行状況等。
以上を踏まえると、本剤の接種対象年齢を16歳以上とすることは可能と考える、と回答した。
「製造販売後の検討事項について」
申請者の説明は、
- 本剤の長期データを含む日本人の安全性について、製造販売承認時までに得られる情報は限定的であり、本剤接種後のSARS-CoV-2感染時に、疾患増強が理論上引き起こされる可能性もあることから、疾患増強等のリスクを含め、本剤2回目接種後12カ月間の安全性を検討することを目的とした使用成績調査( 医療従事者を対象とした先行接種者健康状況調査
(https://www.mhlw.go.jp/content/10906000/ 000721004.pdf(最終確認日:2021年1月21日))での被接種者のうち、本剤2回目接種後12カ月間の追跡調査に同意するすべての被接種者を対象)を実施する予定である。 - また、本剤の臨床試験で十分な安全性情報が得られていない、COVID-19の重症化リスクが高いと考えられる基礎疾患を有する者に対する本剤接種後の安全性の検討を目的として、基礎疾患を有する者を対象とした特定使用成績調査(観察期間:1回目接種日から2回目接種後1カ月)を実施する予定である。
- 国内C4591005試験については、本剤承認後に製造販売後臨床試験に切り替えて長期の安全性等を検討する予定である。
- 本剤の適正使用を促し安全性の確保を図るため、追加のリスク最小化活動として、本剤の副反応集計一覧を一定期間毎に作成し、医療従事者に提供する予定である。
PMDAは、製造販売後調査等の計画に関する申請者の方針は受入れ可能と考える。また、国内の情報のみならず、海外の情報(継続中の海外C4591001試験、海外での使用許可後又は製造販売承認後の情報)を含めて収集し、得られた情報に基づき、本剤の安全性について継続的に評価し、追加の注意喚起や情報提供の要否を検討する等、適切に対応する必要があると考える。
![]()


コメント
Why people still make use of to read news papers
when in this technological world the whole thing is available on net?